

Chronic Lymphocytic Leukemia (CLL) – Sign and Symptoms, Risk Factors, Diagnosis, Complications, Treatment and Prevention
Chronic Lymphocytic Leukemia (CLL) is the most common adult leukemia in Western countries, accounting for approximately 25–30% of all leukemias in the United States and Europe. It is a clonal malignancy of mature-appearing but functionally incompetent CD5+CD19+CD23+ B lymphocytes that accumulate progressively in the peripheral blood, bone marrow, lymph nodes, and spleen due to impaired apoptosis and prolonged cellular survival — rather than primarily from accelerated proliferation. CLL is predominantly a disease of the elderly, with a median age of diagnosis of approximately 70 years, a male predominance (male-to-female ratio approximately 2:1), and is exceedingly rare in individuals under 40 years. Globally, CLL affects approximately 200,000 newly diagnosed patients annually, with the highest incidence rates in North America, Europe, and Australia, and significantly lower rates in East Asia and sub-Saharan Africa — suggesting strong genetic and environmental determinants. 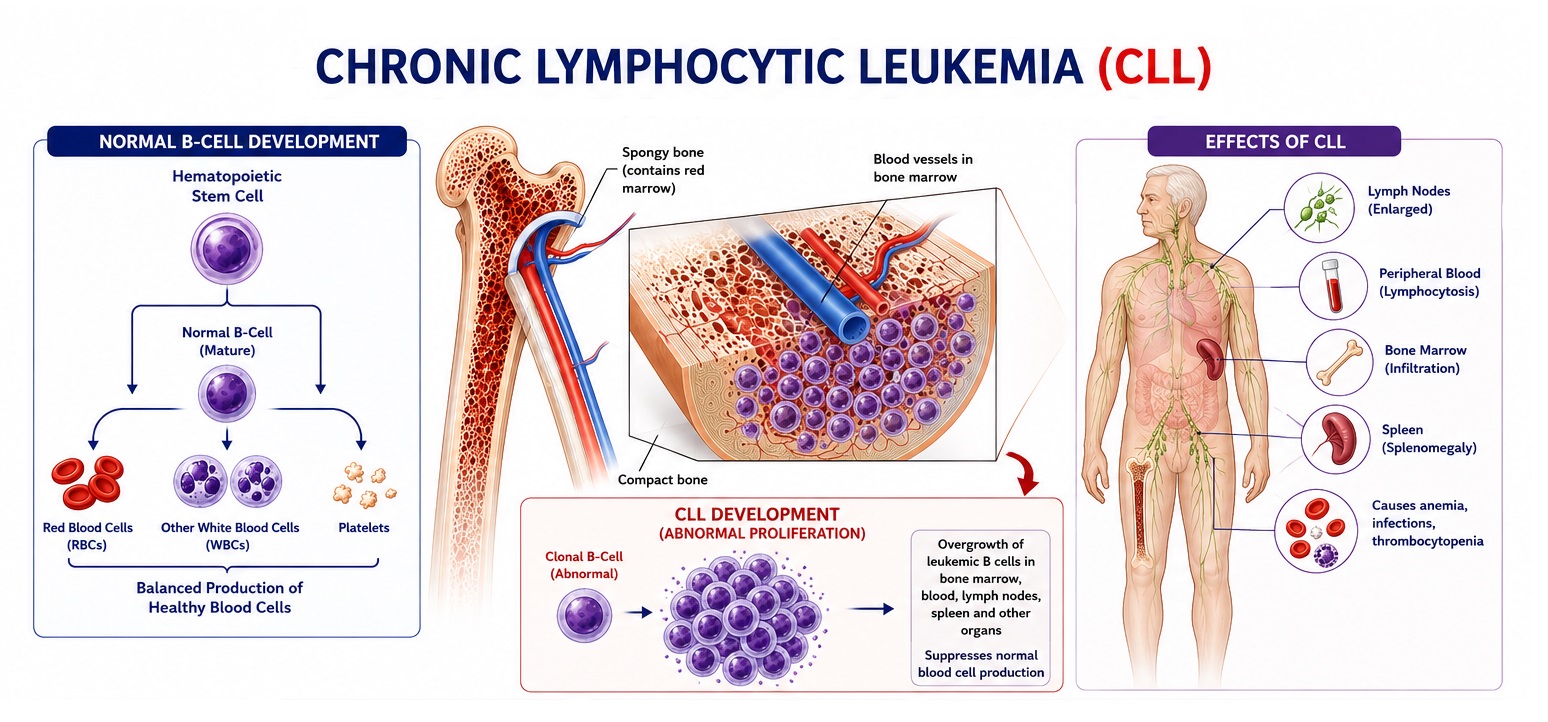
At the molecular level, CLL arises from antigen-experienced B cells that have acquired somatic mutations in immunoglobulin heavy chain variable region genes (IGHV), with IGHV mutational status — unmutated (<2% somatic mutation from germline — U-CLL) versus mutated (M-CLL) — serving as the most powerful single prognostic biomarker. U-CLL originates from naive B cells with unmutated BCR signaling, pursues an aggressive clinical course with shorter time to treatment and inferior overall survival, while M-CLL, arising from post-germinal center B cells, follows an indolent trajectory often compatible with normal life expectancy. Recurrent cytogenetic and molecular abnormalities — including del(13q14) (the most common, ~55%; favorable), del(11q) (~18%; adverse), trisomy 12 (~16%; intermediate), del(17p) (~7%; most adverse) targeting the TP53 tumor suppressor gene, and somatic mutations in NOTCH1, SF3B1, BIRC3, ATM, and TP53 — collectively define the genomic landscape and guide treatment selection.
The clinical presentation of CLL is extraordinarily heterogeneous, ranging from an asymptomatic incidentally detected lymphocytosis in an otherwise healthy individual to an aggressive, symptomatic disease with bulky lymphadenopathy, hepatosplenomegaly, severe cytopenias, recurrent opportunistic infections, and rapid progression. Staging uses the Rai system (United States; stages 0–IV) and Binet system (Europe; stages A–C), both of which are clinical staging systems that stratify patients based on the extent of lymphoid organ involvement and degree of cytopenia. Contemporary integrated prognostic models — including the CLL International Prognostic Index (CLL-IPI) — incorporate clinical stage, IGHV status, TP53 mutation/del(17p), β₂-microglobulin, and age to provide granular individual patient risk stratification.
A paradigmatic shift in CLL management has been catalyzed by the development of highly effective targeted oral therapies. Bruton's tyrosine kinase (BTK) inhibitors — ibrutinib, acalabrutinib, and zanubrutinib — have transformed the treatment landscape by providing durable disease control through sustained BCR signaling suppression, with superior progression-free and overall survival compared to traditional chemoimmunotherapy in multiple randomized trials across all patient subgroups, including those with high-risk genomic features such as del(17p)/TP53 mutation. The BCL-2 inhibitor venetoclax, combined with anti-CD20 antibody obinutuzumab or rituximab, provides time-limited, fixed-duration treatment achieving high rates of complete remission and undetectable minimal residual disease (uMRD) — an emerging endpoint predictive of long-term outcomes. Despite the absence of a proven curative strategy outside allogeneic stem cell transplantation (reserved for young, fit patients with relapsed/refractory high-risk disease), the modern therapeutic era has transformed CLL from a uniformly progressive disease to a manageable chronic condition with excellent long-term outcomes for many patients.
Classification and Staging of CLL
CLL staging guides treatment decisions, follow-up intensity, and trial eligibility. Two clinical staging systems remain the international standard, complemented by modern prognostic indices incorporating molecular and cytogenetic data.
A. Rai Staging System (United States)
B. Binet Staging System (Europe)
| Binet Stage | Criteria | Risk Group | Median Survival |
|---|---|---|---|
| Stage A | Hb ≥10 g/dL; Platelets ≥100×10⁹/L; <3 lymphoid areas involved | Low | >10 years (near-normal life expectancy) |
| Stage B | Hb ≥10 g/dL; Platelets ≥100×10⁹/L; ≥3 lymphoid areas involved | Intermediate | ~5–7 years |
| Stage C | Hb <10 g/dL OR Platelets <100×10⁹/L (due to bone marrow infiltration) | High | ~2–3 years |
Note: The five lymphoid areas counted in Binet staging are: (1) cervical, (2) axillary, (3) inguinal lymph nodes (unilateral or bilateral counted as one), (4) spleen, and (5) liver.
C. CLL-IPI (International Prognostic Index)
CLL-IPI Parameters (Points)
TP53 deletion/mutation: 4 pts
IGHV unmutated: 2 pts
β₂-microglobulin >3.5 mg/L: 2 pts
Rai stage I–IV (or Binet B–C): 1 pt
Age >65 years: 1 pt
CLL-IPI Risk Groups
Score 0–1: Low risk — 93% 5-year OS
Score 2–3: Intermediate risk — 79% 5-year OS
Score 4–6: High risk — 63% 5-year OS
Score 7–10: Very high risk — 23% 5-year OS
(Pre-ibrutinib era data; modern treatment improves outcomes)
Cytogenetic Risk Hierarchy
Favorable: del(13q) sole abnormality
Intermediate: Normal karyotype, Trisomy 12
Adverse: del(11q)
Very Adverse: del(17p), TP53 mutation, complex karyotype (≥3 unrelated abnormalities)
Signs and Symptoms of Chronic Lymphocytic Leukemia
CLL presents across a remarkably broad clinical spectrum — from asymptomatic incidental discovery of lymphocytosis on a routine blood count to a rapidly progressive, symptomatic disease with systemic features of advanced malignancy. The majority (~70–80%) of CLL patients are diagnosed incidentally in the asymptomatic early stage during evaluation for an unrelated complaint. Symptom onset reflects increasing disease burden, impaired hematopoiesis, immune dysregulation, and organ infiltration by clonal B lymphocytes.
Key Clinical Concept — "B Symptoms" in CLL: The presence of constitutional B symptoms (fever >38°C without infection, drenching night sweats, unintentional weight loss >10% body weight over 6 months) in CLL indicates progressive, biologically aggressive disease and is one of the standard indications triggering initiation of treatment per iwCLL 2018 guidelines. 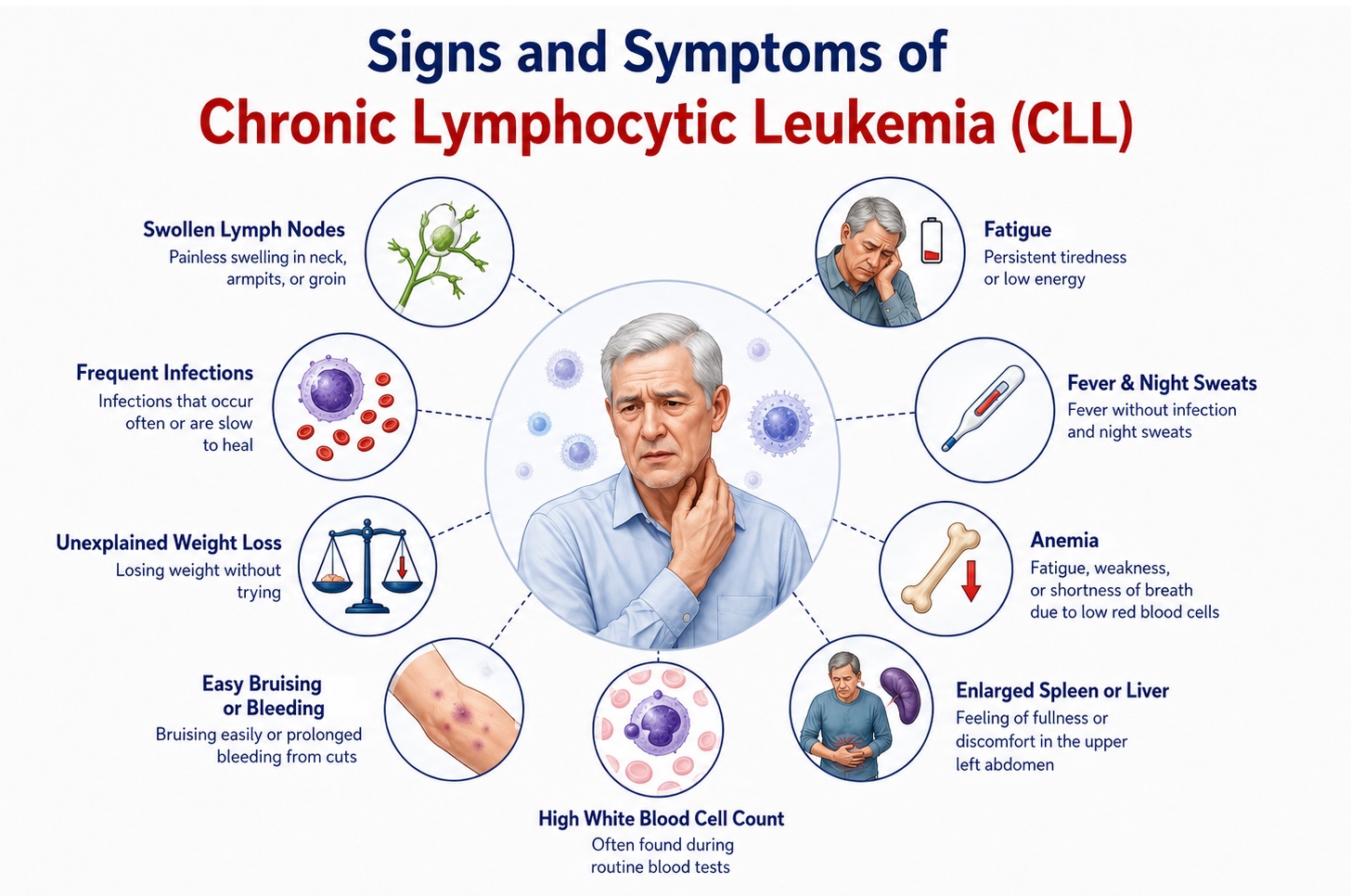 1. Constitutional and Systemic Symptoms
1. Constitutional and Systemic Symptoms
- Fatigue and profound weakness: The most common and debilitating symptom in symptomatic CLL, reported by approximately 80% of patients requiring treatment. Fatigue arises from multifactorial causes: anemia from bone marrow infiltration by leukemic lymphocytes replacing erythroid precursors, tumor cytokine burden (TNF-α, IL-6), impaired sleep from night sweats, deconditioning, and the psychological burden of a chronic malignant diagnosis. Fatigue in CLL is frequently disproportionate to the measurable degree of anemia and may persist even after disease response to treatment.
- Unintentional weight loss: Progressive weight loss exceeding 10% of body weight over 6 months reflects the hypermetabolic state of advancing CLL, where rapidly proliferating leukemic clones in proliferation centers (pseudofollicles) within lymph nodes generate continuous metabolic demands, elevated circulating cytokines suppress appetite, and tumor-related cachexia promotes muscle catabolism. Significant weight loss is a treatment indication per iwCLL guidelines.
- Fever without apparent infection: Low-grade fever occurring in the absence of identified infection may reflect tumor-associated cytokine release, particularly interleukin-6 and tumor necrosis factor-alpha. However, given the profound immunodeficiency of CLL (hypogammaglobulinemia, T-cell dysfunction), infection must always be actively excluded as the primary etiology — fever in CLL demands urgent microbiological workup.
- Drenching night sweats: Profuse nocturnal perspiration saturating bedclothes is a hallmark B symptom of CLL, attributable to autonomic dysregulation, cytokine-mediated thermoregulatory disturbance, and tumor necrosis factor activity. Night sweats signal disease progression requiring evaluation for treatment initiation or Richter transformation.
- Reduced exercise tolerance and exertional dyspnea: Diminishing functional capacity correlates with advancing anemia, splenomegaly impairing diaphragmatic excursion, pleural effusion from lymphomatous infiltration, and the general deconditioning accompanying chronic malignant disease. Dyspnea may also indicate pulmonary parenchymal infiltration by CLL cells — a rare but recognized manifestation.
2. Lymphadenopathy
- Peripheral lymphadenopathy: Painless, rubbery, non-tender, discrete, and mobile enlarged lymph nodes represent one of the most frequent clinical findings in CLL, detected in approximately 60–70% of patients at diagnosis. Nodes are typically bilateral and symmetrical, affecting the cervical chain most prominently, followed by axillary and inguinal regions. Unlike reactive or infectious lymphadenopathy, CLL-associated nodes are firm-to-rubbery in consistency, non-erythematous, and non-tender — though rapid enlargement or constitutional B symptoms alongside lymph node changes raise concern for Richter transformation to aggressive large cell lymphoma.
- Mediastinal and retroperitoneal lymphadenopathy: Internal lymph node enlargement detected on CT imaging — not clinically palpable — may cause symptoms through mass effect: mediastinal nodes producing superior vena cava syndrome (facial/neck swelling, venous engorgement, dyspnea), hilar nodes causing bronchial compression (persistent cough, wheeze, post-obstructive pneumonia), and retroperitoneal nodes producing hydronephrosis, ureteral obstruction, or bowel compression symptoms.
- Waldeyer's ring involvement: Enlargement of the palatine tonsils, adenoids, and lingual tonsil as part of CLL infiltration, which may cause obstructive sleep apnea, dysphagia, and recurrent oropharyngeal infections.
3. Splenomegaly and Hepatomegaly
- Splenomegaly: Splenic enlargement occurs in approximately 40–50% of CLL patients due to leukemic infiltration of the red pulp and white pulp of the spleen by accumulating B lymphocytes. Clinical manifestations include left upper quadrant dull aching pain or heaviness, early satiety from gastric compression by the enlarged organ, and abdominal fullness. Massive splenomegaly may cause mechanical complications including acute splenic infarction (sudden severe left upper quadrant pain radiating to the left shoulder), thrombocytopenia from hypersplenism (platelet sequestration in the enlarged spleen), and hemolytic anemia from accelerated erythrocyte destruction. Splenic rupture, though rare, represents a potentially life-threatening complication requiring emergency splenectomy.
- Hepatomegaly: Hepatic infiltration by CLL lymphocytes occurs in approximately 30–40% of patients, typically producing mild, non-tender hepatomegaly detectable clinically and on imaging. Rarely, periportal infiltration produces cholestatic jaundice or hepatic dysfunction mimicking portal hypertension. Hepatomegaly is included in the Binet and Rai staging systems as a criterion of organ involvement.
4. Features of Bone Marrow Infiltration and Cytopenias
- Anemia: Normochromic normocytic anemia develops from progressive replacement of normal erythroid hematopoiesis by infiltrating CLL cells in the bone marrow — reducing erythropoietin-responsive erythroid precursors — and from concurrent immune-mediated autoimmune hemolytic anemia (AIHA), a CLL-specific complication that may dominate the hematological picture. Patients present with progressive pallor, fatigue, dyspnea on exertion, and palpitations. AIHA in CLL is characterized by positive direct antiglobulin test (DAT/Coombs), reticulocytosis, elevated indirect bilirubin, elevated LDH, and low haptoglobin. Hemoglobin below 11 g/dL due to bone marrow infiltration (excluding immune causes) constitutes a treatment indication.
- Thrombocytopenia: Reduced platelet count arising from bone marrow failure (marrow replaced by CLL cells reducing megakaryopoiesis) or immune-mediated immune thrombocytopenic purpura (ITP — a specific autoimmune complication of CLL) causing peripheral platelet destruction. Clinical manifestations include petechiae, purpura, ecchymoses, mucosal bleeding (gingival, epistaxis, menorrhagia), and — in severe thrombocytopenia — spontaneous intracranial hemorrhage. Platelet count below 100 × 10⁹/L due to bone marrow failure constitutes a treatment indication per iwCLL guidelines.
- Neutropenia: Impaired neutrophil production from marrow infiltration, exacerbated by chemoimmunotherapy-induced myelosuppression during treatment, predisposes to bacterial infections. Unlike the profound neutropenia of acute leukemias, CLL-associated neutropenia is typically more modest but clinically significant, particularly in the context of concurrent T-cell dysfunction and hypogammaglobulinemia.
5. Immunodeficiency and Infectious Manifestations
Immunodeficiency is a fundamental and clinically critical feature of CLL, arising from multiple overlapping mechanisms: hypogammaglobulinemia (reduced IgG, IgA, IgM production), T-cell numerical and functional deficits, complement deficiency, neutropenia, and CLL-induced immune checkpoint upregulation suppressing anti-tumor immune surveillance. Infectious complications are the leading cause of death in CLL, responsible for approximately 50% of all CLL-associated mortality.
- Hypogammaglobulinemia and recurrent bacterial infections: Progressive decline in all serum immunoglobulin classes occurs in approximately 50–70% of CLL patients, with severity correlating with advancing disease stage and prior treatment. Encapsulated bacterial pathogens — Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus — are the predominant organisms causing recurrent sinopulmonary infections (chronic sinusitis, bronchitis, pneumonia, otitis media) and bacteremia. Immunoglobulin replacement therapy is used in patients with recurrent infections and documented hypogammaglobulinemia.
- Viral infections and reactivation: T-cell dysfunction in CLL impairs viral immune surveillance, predisposing to reactivation of latent viral infections. Herpes zoster (shingles) occurs at approximately 10-fold the frequency of the age-matched general population and may be disseminated or ophthalmic. Herpes simplex virus causes severe, atypical, slow-healing mucocutaneous ulcers. Cytomegalovirus (CMV), Epstein-Barr virus (EBV), and JC virus reactivation occur particularly during intensive treatment with anti-CD20 antibodies, fludarabine, or Bruton's tyrosine kinase inhibitors. Progressive multifocal leukoencephalopathy (PML) from JC virus reactivation is a rare but devastating neurological complication.
- Opportunistic infections: In advanced or heavily treated CLL, Pneumocystis jirovecii pneumonia (PCP), invasive aspergillosis, Cryptococcus neoformans meningitis, and Mycobacterium tuberculosis or non-tuberculous mycobacteria may occur, reflecting profound acquired immunosuppression. PCP prophylaxis with trimethoprim-sulfamethoxazole is recommended during and after intensive chemoimmunotherapy.
- COVID-19 and poor vaccine response: CLL patients demonstrate profoundly impaired humoral responses to SARS-CoV-2 vaccination, achieving protective antibody titers in only 30–50% of cases — with the worst responses observed in those on BTK inhibitors or with prior rituximab therapy. COVID-19 causes disproportionate morbidity and mortality in CLL, with higher rates of severe disease, prolonged viral shedding, and secondary bacterial pneumonia. Pre-exposure prophylaxis with long-acting monoclonal antibodies (tixagevimab-cilgavimab) and early antiviral therapy (nirmatrelvir-ritonavir) are recommended for CLL patients.
6. Autoimmune Complications
- Autoimmune hemolytic anemia (AIHA): Occurs in approximately 7–10% of CLL patients, mediated by IgG warm autoantibodies (warm AIHA) — or less commonly cold agglutinins (IgM) — produced by non-malignant bystander B cells dysregulated by the CLL microenvironment. AIHA may be precipitated by fludarabine-containing chemotherapy and is characterized by hemolytic jaundice, dark urine, pallor, fatigue, and positive DAT. Treatment involves corticosteroids, rituximab, IV immunoglobulin, and addressing the underlying CLL. AIHA dramatically worsens anemia beyond what bone marrow infiltration alone produces.
- Immune thrombocytopenic purpura (ITP): Occurring in ~2–5% of CLL patients, ITP involves autoantibody-mediated destruction of platelets, presenting clinically indistinguishable from bone marrow failure-related thrombocytopenia. Distinguishing ITP (peripheral destruction; responds to immunosuppression) from bone marrow failure (central production failure; requires CLL treatment) is critical and necessitates bone marrow examination to assess megakaryocytic adequacy.
- Pure red cell aplasia (PRCA): A rare autoimmune complication in which antibodies (or cytotoxic T cells) target erythroid precursors in the bone marrow, producing profound erythroid aplasia with severe anemia, absent reticulocytes, and absent bone marrow erythroid precursors in the setting of normal megakaryocytes and myeloid cells. Treatment involves cyclosporine, glucocorticoids, and rituximab.
- Autoimmune neutropenia: Least common autoimmune cytopenia in CLL; antibody-mediated neutrophil destruction compounds chemotherapy-induced neutropenia, increasing infectious risk.
7. Skin Manifestations
- Leukemia cutis: Specific cutaneous infiltration by CLL B lymphocytes produces flesh-colored to violaceous papules, nodules, or plaques — most commonly on the face, trunk, and extremities. Biopsy confirms CLL infiltration and distinguishes leukemia cutis from reactive skin conditions.
- Insect bite hypersensitivity: An unusual and characteristic immune complication of CLL — particularly in East Asian patients and those with EBV co-infection — manifesting as severe, exaggerated local reactions (extensive edema, vesiculation, ulceration, necrosis) to mosquito bites, accompanied by systemic symptoms (fever, lymphadenopathy, NK/T-cell lymphoma-like features). Likely reflects EBV-driven immune dysregulation in CLL T and NK cells.
- Dermatological infections: Herpes zoster producing dermatomal or disseminated vesicular rash, severe herpes simplex ulceration, and fungal skin infections (tinea, candidiasis) are frequent cutaneous manifestations of CLL-associated immunodeficiency.
- Secondary skin malignancies: CLL-associated immunosuppression dramatically increases the risk of skin cancers — particularly squamous cell carcinoma (SCC), which follows an accelerated and more aggressive course in CLL patients, with higher rates of local recurrence, metastasis, and SCC-related death. Regular dermatological surveillance is recommended.
8. Signs on Physical Examination
- Peripheral lymphadenopathy: Bilateral, painless, rubbery enlarged cervical, axillary, and inguinal lymph nodes — the most common objective clinical finding in CLL, appreciated on systematic lymph node examination.
- Splenomegaly: Detectable on abdominal percussion (Traube's space dullness) and palpation; massive splenomegaly may extend to the right iliac fossa. Splenic rub suggests infarction.
- Hepatomegaly: Smooth, non-tender hepatic enlargement on percussion and palpation below the right costal margin.
- Pallor: Conjunctival and palmar pallor reflecting anemia from marrow infiltration or AIHA.
- Petechiae and purpura: Non-blanching cutaneous hemorrhages from thrombocytopenia.
- Jaundice: Scleral icterus and jaundice in AIHA (hemolytic jaundice) or rare hepatic infiltration with cholestasis.
- Sternal and bony tenderness: Mild bony tenderness on percussion of the sternum and ribs from medullary expansion by leukemic infiltration — less prominent than in acute leukemias.
Risk Factors of Chronic Lymphocytic Leukemia
CLL has one of the strongest genetic predispositions of any hematological malignancy, with environmental factors playing a relatively minor role compared to inherited susceptibility. The precise etiopathogenesis involves an interaction between antigen-driven B-cell stimulation, germline susceptibility loci, and acquired somatic mutations promoting clonal B-lymphocyte expansion and survival. 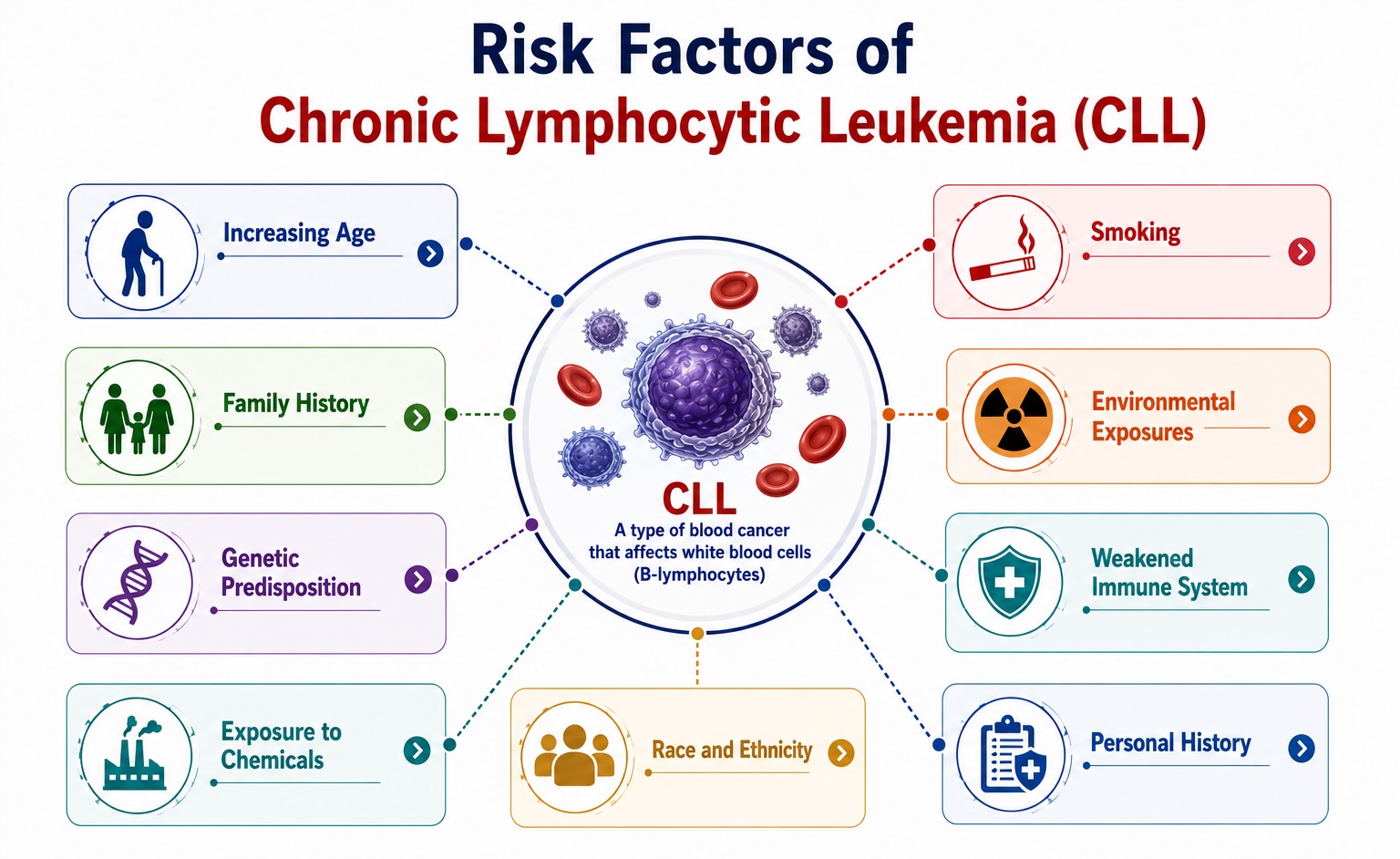
1. Genetic and Hereditary Risk Factors
- Positive family history of CLL or B-cell malignancies: The most potent known CLL risk factor — first-degree relatives of CLL patients have an approximately 7–9-fold increased risk of developing CLL compared to the general population. This familial aggregation is the highest of any leukemia subtype. In large familial CLL kindreds, disease typically presents at an earlier age and with higher disease severity in successive generations. First-degree relatives also demonstrate an increased prevalence of monoclonal B-cell lymphocytosis (MBL) — the pre-malignant precursor state — at rates of 13–18% compared to 3–5% in the general population.
- Germline susceptibility variants (GWAS-identified loci): Genome-wide association studies have identified over 40 common germline susceptibility variants associated with CLL risk, including single nucleotide polymorphisms (SNPs) in or near genes encoding IRF4 (lymphoid differentiation transcription factor), LEF1, RFX7, POT1, PMAIP1/NOXA, HLA region genes, FCRL2, BAX, and BCL2 family members — collectively accounting for approximately 15–20% of familial CLL risk. No single high-penetrance Mendelian gene (analogous to BRCA1/2 in breast cancer) has been identified for CLL.
- Monoclonal B-cell lymphocytosis (MBL): MBL — defined as the presence of a monoclonal B-cell population <5 × 10⁹/L without lymphadenopathy, splenomegaly, cytopenias, or clinical features of CLL — is the obligate precursor state of CLL. Population-based studies identify MBL in approximately 3–5% of individuals over 40 years and 8–15% of first-degree relatives of CLL families. Low-count MBL (<0.5 × 10⁹/L) carries minimal malignant potential; high-count MBL (0.5–5 × 10⁹/L) progresses to CLL requiring treatment at a rate of approximately 1–2% per year — establishing a model analogous to MGUS in multiple myeloma.
- Ethnic and racial predisposition: CLL predominantly affects individuals of Northern European Caucasian ancestry, with incidence rates 5–10-fold lower in East Asians (Chinese, Japanese, Korean) even in immigrant populations maintaining Western lifestyles — strongly suggesting inherent genetic population-level differences in CLL susceptibility rather than purely environmental causation. African Americans have intermediate CLL incidence but tend to present with more aggressive disease features. CLL is rare in South Asia, sub-Saharan Africa, and Latin America.
2. Demographic Risk Factors
- Age: The risk of CLL increases exponentially with advancing age. Median age at diagnosis is approximately 70–72 years; CLL below age 40 is exceptionally rare (<1% of cases). The age-related increase reflects accumulation of somatic mutations in B lymphocytes, age-associated changes in immune regulation permitting clonal expansion, and the natural history of MBL-to-CLL progression over years to decades.
- Male sex: Males are affected approximately twice as frequently as females (male-to-female ratio ~2:1), consistent across geographic populations and ethnic groups. Possible explanations include differences in hormonal milieu (estrogens may have protective immunological effects), occupational exposures, and X-chromosome-linked genetic differences in immune regulation. Female CLL patients tend to have better overall survival than males with comparable disease, partly attributable to less aggressive disease biology and potentially differential hormonal immune effects.
3. Environmental and Occupational Exposures
- Agent Orange / Herbicide exposure: Exposure to herbicides — particularly organochlorine pesticides and Agent Orange (dioxin-containing herbicide used during the Vietnam War) — has been most consistently linked to increased CLL risk. The United States Department of Veterans Affairs recognizes CLL as a service-connected disability for Vietnam veterans with documented Agent Orange exposure, acknowledging a causal relationship. Dioxins promote B-cell proliferation through aryl hydrocarbon receptor (AHR) signaling and impair normal lymphocyte apoptosis.
- Pesticides and agricultural chemicals: Multiple epidemiological studies demonstrate positive associations between CLL risk and occupational or residential exposure to organophosphate pesticides, carbamate insecticides, and phenoxyacid herbicides in agricultural workers, pesticide manufacturers, and residents of farming communities. The strength of association is moderate, with relative risks of 1.3–2.0 in exposed versus unexposed populations.
- Ionizing radiation: Unlike AML, the evidence for ionizing radiation as a CLL risk factor is inconsistent and weaker. Atomic bomb survivor data suggest a smaller radiation-attributable excess risk for CLL than for other leukemias, consistent with the hypothesis that CLL is fundamentally a disease of antigen-driven B-cell stimulation rather than primarily DNA damage-driven leukemogenesis. Occupational radiation exposure in radiologists and nuclear workers has been associated with modestly elevated CLL risk in some studies.
- Hair dye use: Long-term personal use of permanent oxidative hair dyes — containing aromatic amines with potential genotoxicity — has been associated with a modestly increased CLL risk in several case-control studies, with stronger associations for darker colors and longer duration of use. However, the association is less robust than for non-Hodgkin lymphoma more broadly.
- Viral infections: Chronic antigen stimulation from viral pathogens — particularly hepatitis C virus (HCV) — has been implicated in CLL pathogenesis through persistent B-cell receptor stimulation promoting clonal B-cell expansion. HCV eradication with direct-acting antivirals has been reported to produce CLL responses in rare cases. EBV, HTLV-1, and SV40 have been investigated but their causal roles in CLL remain unestablished.
4. Immune and Medical Risk Factors
- Personal history of autoimmune disease: A history of autoimmune conditions — including rheumatoid arthritis, systemic lupus erythematosus, and Sjögren's syndrome — is associated with increased CLL risk, possibly reflecting shared pathways of chronic immune dysregulation, abnormal B-cell homeostasis, and immune checkpoint dysfunction that predispose to B-cell lymphoproliferative transformation.
- Prior blood transfusions: Some epidemiological studies report a modestly increased CLL risk following blood transfusions, possibly through alloantigen-driven immune stimulation or transfusion-related viral exposures, though evidence remains inconsistent.
Diagnosis of Chronic Lymphocytic Leukemia
Diagnosis of CLL requires integration of clinical findings, peripheral blood count and morphology, immunophenotyping, cytogenetics, and molecular profiling. According to the iwCLL 2018 diagnostic criteria, CLL is defined by the co-presence of: (1) peripheral blood B-lymphocyte count ≥5 × 10⁹/L of monoclonal B cells, (2) persistence for ≥3 months, and (3) a characteristic CLL immunophenotype — without requirement for bone marrow biopsy or lymph node biopsy for diagnosis in most cases.
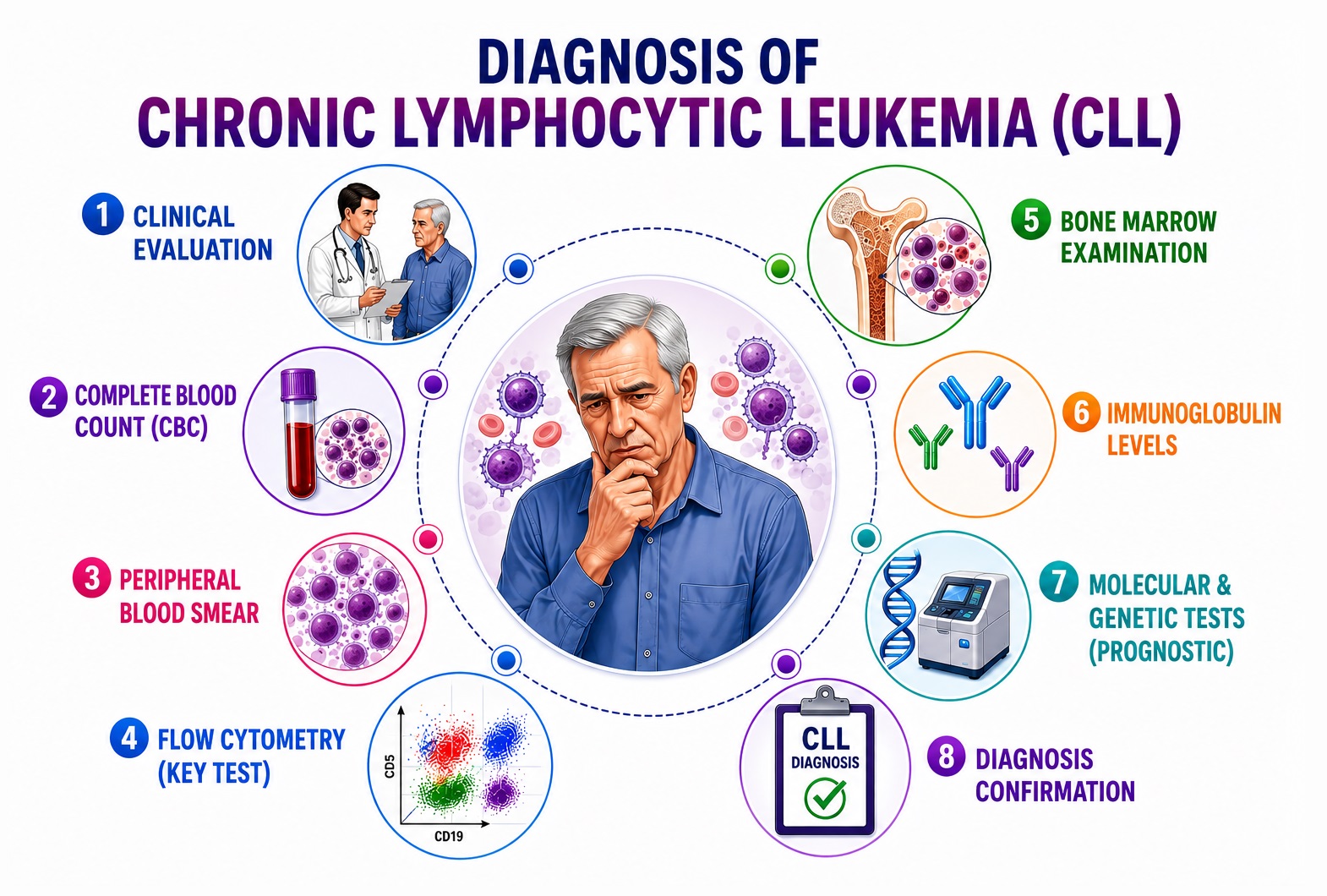
1. Clinical Evaluation
- Symptom assessment: Constitutional B symptoms (fever, night sweats, weight loss >10%), fatigue severity, lymph node changes noticed by the patient (size, rate of change, tenderness), abdominal symptoms (early satiety, left upper quadrant discomfort suggesting splenomegaly), and infectious history (recurrent sinopulmonary infections suggesting hypogammaglobulinemia, herpes zoster episodes).
- Family history: Detailed history of hematological malignancies — particularly CLL, non-Hodgkin lymphoma, Waldenström macroglobulinemia — in first- and second-degree relatives; may influence germline genetic testing decisions in young patients or those with strong family history.
- Medication and prior therapy history: Prior immunosuppressive therapy, chemotherapy for other malignancies, steroid use (relevant to infection risk), and drug-drug interaction potential for targeted therapies (BTK inhibitors have multiple interactions with CYP3A4 substrates and anticoagulants).
- Performance status assessment: ECOG/WHO performance status is critical for treatment eligibility classification — "fit" (CIRS score ≤6, creatinine clearance ≥70 mL/min) versus "unfit" (CIRS score >6 or creatinine clearance <70 mL/min) determination guides treatment selection, as intensive chemoimmunotherapy is generally restricted to fit patients.
- Systematic lymph node examination across all accessible regions (cervical — anterior and posterior chains, supraclavicular, axillary, epitrochlear, inguinal, popliteal); documentation of size, number, consistency, and symmetry
- Abdominal examination: hepatic and splenic size measurement in centimeters below costal margins; splenic tenderness suggesting infarction
- Skin examination: leukemia cutis, infection evidence (herpes zoster, fungal infections), signs of thrombocytopenia (petechiae, purpura), pallor of anemia, jaundice of AIHA
- Cardiovascular examination: relevant for treatment eligibility (anthracycline suitability) and BTK inhibitor cardiac risk assessment (atrial fibrillation risk)
2. Peripheral Blood Analysis
- Lymphocytosis: Absolute lymphocyte count (ALC) ≥5 × 10⁹/L — the defining quantitative criterion for CLL diagnosis. ALC may range from 5 to over 500 × 10⁹/L in different patients; the rate of lymphocyte doubling time (LDT) — the time for the ALC to double — is a clinically useful prognostic indicator, with LDT <12 months indicating more aggressive disease biology.
- Peripheral blood smear morphology: The characteristic cytological appearance of CLL is mature-appearing small lymphocytes with scant cytoplasm, condensed "soccer ball" (clumped) chromatin, and inconspicuous nucleoli — reflecting the arrested maturation of CLL B cells at the mature B-cell stage. A critical pathognomonic finding is smudge cells (basket cells or Gumprecht shadows) — ruptured lymphocytes that, due to the fragile cytoskeleton of CLL cells, smear during slide preparation, leaving ghostly nuclear remnants without cytoplasmic borders. Smudge cells are virtually pathognomonic of CLL on blood smear and distinguish it from other lymphocytoses. Prolymphocytes, large lymphocytes with prominent nucleoli, should comprise <10% of lymphoid cells; >55% prolymphocytes defines B-cell prolymphocytic leukemia (B-PLL), a distinct and more aggressive entity.
- Anemia: Normochromic normocytic anemia from marrow infiltration; hemolytic pattern (elevated MCV, elevated reticulocyte count, elevated unconjugated bilirubin, elevated LDH, reduced haptoglobin) in concurrent AIHA.
- Thrombocytopenia: Varying severity, from mild (>100 × 10⁹/L) to severe (<20 × 10⁹/L) depending on degree of marrow infiltration or concurrent ITP.
- Neutropenia: Mild to moderate reduction in absolute neutrophil count from marrow infiltration; severe neutropenia after chemoimmunotherapy.
3. Immunophenotyping (Flow Cytometry)
Multiparameter flow cytometry immunophenotyping of peripheral blood is the gold standard for CLL diagnosis and differential diagnosis from other B-cell lymphoproliferative disorders:
| Marker | CLL | MCL | Follicular Lymphoma | Hairy Cell Leukemia |
|---|---|---|---|---|
| CD5 |  Positive Positive |
Positive |
 Negative Negative |
Negative |
| CD19 | Positive |
Positive |
Positive |
Positive |
| CD23 | Positive |
Negative |
Variable | Negative |
| CD20 | Dim (weak) | Bright | Bright | Bright |
| FMC7 | Negative |
Positive |
Positive |
Positive |
| Surface Ig | Dim (weak) | Bright | Bright | Bright |
| CD79b | Dim | Bright | Bright | Dim |
| CD103 | Negative |
Negative |
Negative |
Positive |
| Cyclin D1 | Negative |
Positive |
Negative |
Negative |
| Matutes Score | 4–5 (diagnostic) | ≤2 | 1–2 | 1–2 |
The Matutes scoring system assigns one point each for CD5 positivity, CD23 positivity, weak/absent surface Ig, weak/absent CD79b, and negative FMC7 — a score of 4 or 5 is diagnostic of CLL with high specificity.
4. Bone Marrow Examination
- Bone marrow aspiration: Not required for diagnosis of CLL per iwCLL 2018 criteria but performed pre-treatment to assess marrow infiltration pattern and degree, evaluate cytopenia etiology, and establish a baseline for MRD monitoring during treatment.
- Trephine biopsy histology: Characterizes the marrow infiltration pattern — nodular (best prognosis), interstitial, mixed, or diffuse (worst prognosis) — with diffuse infiltration correlating with advanced disease and poor treatment response. CLL cells appear as monotonous small lymphocytes with immunohistochemical positivity for CD20, CD5, CD23, CD43, and BCL-2.
- Marrow infiltration percentage: Normal marrow cellularity displaced by ≥30% CLL lymphocytes is typical; advanced disease may show near-complete marrow replacement. The degree of marrow infiltration influences the risk of treatment-emergent cytopenias and the depth of remission achievable.
5. Cytogenetic Analysis
FISH analysis of peripheral blood or bone marrow is the preferred cytogenetic method in CLL, detecting the major prognostically relevant abnormalities in non-dividing CLL cells (which rarely enter mitosis spontaneously, limiting conventional karyotyping sensitivity). The standard CLL FISH panel includes:
- del(13q14) — most common (~55%): Deletion of the miR-15a/miR-16-1 microRNA cluster at 13q14 removes post-transcriptional BCL-2 repressors, enhancing CLL cell survival. Sole del(13q) is the most favorable cytogenetic finding — associated with the longest median time to treatment and best overall survival, particularly in patients with mutated IGHV.
- del(11q22–23) — ATM deletion (~18%): Loss of the ataxia-telangiectasia mutated (ATM) gene at chromosome 11q impairs double-strand DNA break repair and p53-mediated apoptotic response, conferring accelerated disease progression, bulky lymphadenopathy, and inferior outcomes with alkylator-based chemotherapy. Responds well to BTK inhibitors.
- Trisomy 12 (~16%): Gain of an extra copy of chromosome 12 produces intermediate prognosis; associated with atypical CLL morphology and increased frequency of NOTCH1 mutations, CD38 positivity, and potential for transformation to DLBCL.
- del(17p13.1) — TP53 deletion (~7%; rises to ~25–30% in relapsed/refractory): Loss of one TP53 allele, typically combined with somatic mutation of the remaining allele (biallelic TP53 inactivation), abrogates genotoxic stress-induced apoptosis — rendering CLL cells completely resistant to conventional alkylating agents and purine analogues. del(17p)/TP53 mutation defines the highest-risk cytogenetic category, with aggressive disease progression, very short time to treatment, resistance to chemoimmunotherapy, and short overall survival with conventional treatment. BTK inhibitors and venetoclax-based regimens overcome TP53-mediated drug resistance and are the treatment of choice.
- Complex karyotype (≥3 unrelated chromosomal abnormalities): Associated with poor outcomes across treatment modalities; may require stimulated karyotyping with CpG oligonucleotide plus IL-2 culture conditions to induce CLL cell division.
6. Molecular Testing
- IGHV mutation status: Sequencing of the immunoglobulin heavy chain variable region gene determines whether CLL has unmutated (<2% deviation from germline sequence — U-CLL; poor prognosis, shorter time to treatment, more aggressive disease) or mutated (≥2% deviation — M-CLL; favorable prognosis, longer indolent phase, longer survival) IGHV. IGHV 3-21 gene usage, regardless of mutational status, confers an intermediate-poor prognosis ("stereotyped subset 2") and should be specifically reported. IGHV status is the most powerful independent prognostic biomarker in CLL and guides treatment expectations, though not individual drug selection per current guidelines.
- TP53 mutation analysis (next-generation sequencing): Mandatory before initiating any CLL treatment. TP53 mutations (exons 4–8 of the TP53 gene) may occur without concurrent del(17p) (FISH-negative, mutation-positive cases — approximately 5% of newly diagnosed CLL) and are equally clinically significant. Identification of TP53 mutation/del(17p) mandates use of BTK inhibitor or venetoclax-based therapy and avoidance of chemoimmunotherapy.
- NOTCH1 mutations: Present in approximately 8–12% of CLL at diagnosis, rising to 20–25% at relapse. NOTCH1 mutations activate the Notch signaling pathway, promoting CLL cell survival and proliferation, and are associated with unmutated IGHV, trisomy 12, aggressive disease course, and predisposition to Richter transformation. NOTCH1 mutation is an independent adverse prognostic marker.
- SF3B1 mutations: Spliceosome component mutations present in approximately 10–15% of CLL, associated with del(11q), unmutated IGHV, fludarabine refractoriness, and adverse prognosis independent of other factors. SF3B1 mutations generate aberrant RNA splicing patterns contributing to epigenetic dysregulation and enhanced proliferative signaling.
- BIRC3 mutations: Mutations in this NF-κB pathway inhibitor gene occur in approximately 4% of newly diagnosed CLL, rising significantly in relapsed disease. Associated with adverse prognosis comparable to del(11q) and resistance to fludarabine-based therapy.
- ATM mutations (without del(11q)): Somatic mutations in the non-deleted ATM allele in del(11q) patients produce biallelic ATM inactivation — equivalent to del(17p) in terms of DNA damage repair deficiency and poor prognosis.
7. Serum Biomarkers
- β₂-microglobulin (β₂M): Serum level of this cell surface MHC class I-associated protein reflects CLL tumor burden and cellular proliferation rate. β₂M >3.5 mg/L is incorporated into the CLL-IPI as an adverse prognostic parameter and is independently associated with shorter time to treatment and inferior overall survival. Level correlates with disease stage and treatment response.
- Serum LDH: Elevated LDH reflects increased cellular proliferation and turnover; marked elevation raises concern for Richter transformation to diffuse large B-cell lymphoma.
- CD38 expression: CD38 positivity on >30% of CLL cells (by flow cytometry) is an adverse prognostic marker associated with unmutated IGHV, active BCR signaling, and inferior time to treatment — though CD38 expression may be dynamic and not reliably stable throughout the disease course.
- ZAP-70 expression: Aberrant expression of the T-cell-associated tyrosine kinase ZAP-70 in CLL B cells (normally absent) correlates with unmutated IGHV status, enhanced BCR signaling, and adverse prognosis. Less standardized than IGHV sequencing due to technical variability in flow cytometric assessment.
- Serum immunoglobulin levels (IgG, IgA, IgM): Assessment of hypogammaglobulinemia severity guides decision-making for immunoglobulin replacement therapy in patients with recurrent bacterial infections.
- Direct antiglobulin test (DAT/Coombs): Positive in approximately 25–35% of CLL patients at some point during disease course; actively positive with hemolytic anemia confirming AIHA.
- Serum uric acid and electrolytes: Baseline assessment for tumor lysis syndrome prophylaxis planning before treatment initiation, particularly with venetoclax (structured tumor lysis prophylaxis is mandatory during ramp-up dosing).
8. Imaging Studies
- CT scan (neck, chest, abdomen, pelvis): Provides comprehensive assessment of lymphadenopathy distribution and burden (total lymph node size — sum of products of diameters for response assessment), spleen and liver dimensions, and identification of bulky disease. Mandatory in suspected Richter transformation (marked CT metabolic avidity on PET-CT and marked LDH elevation). Not recommended for routine monitoring in asymptomatic early CLL due to cumulative radiation exposure and cost.
- PET-CT scan: Not routinely indicated in CLL (which is typically FDG-low-avidity) but highly valuable for Richter transformation detection — areas of intense FDG-avidity (SUVmax typically >5) correlate with sites of DLBCL transformation and guide biopsy of the highest-avidity node to ensure sampling of transformed disease rather than residual CLL. SUVmax >10 in a CLL patient has a high positive predictive value for Richter transformation.
- Ultrasound: Monitoring of lymphadenopathy and organomegaly in clinical follow-up; preferred over CT for serial assessment due to absence of radiation, availability, and cost.
- MRI brain: Indicated for evaluation of suspected CNS involvement (rare in CLL — headache, focal neurological deficits, cranial nerve palsies, meningismus), Richter transformation with CNS disease, or PML from JC virus reactivation (characteristic white matter lesion pattern on MRI).
9. Minimal Residual Disease (MRD) Assessment
MRD measurement — quantifying residual leukemic burden below morphological detection thresholds — has emerged as a critical endpoint in clinical trials and is increasingly informing treatment duration decisions in clinical practice:
- Multi-color flow cytometry (8–10 color panels): Detects CLL cells at a sensitivity of 10⁻⁴ to 10⁻⁵ (one CLL cell per 10,000–100,000 leukocytes). The EuroFlow 4-color MRD panel (CD19/CD5/CD20/CD38 gating strategy) provides standardized uMRD assessment at 10⁻⁴ sensitivity.
- Allele-specific oligonucleotide PCR (ASO-PCR): Patient-specific IGHV-D-J rearrangement PCR primers detect CLL cells at 10⁻⁵ to 10⁻⁶ sensitivity — the highest available sensitivity for MRD assessment in CLL.
- Next-generation sequencing (NGS) of IGHV-D-J: ClonoSEQ assay and similar platforms provide sensitive, standardized MRD assessment at 10⁻⁵ to 10⁻⁶ sensitivity with broad clinical applicability independent of specific IGHV rearrangement clonotype.
- MRD thresholds: Undetectable MRD (uMRD) is defined as <1 CLL cell per 10,000 leukocytes (10⁻⁴); uMRD is strongly associated with prolonged progression-free and overall survival and increasingly guides decisions regarding treatment duration with fixed-duration venetoclax-based regimens.
Complications of Chronic Lymphocytic Leukemia
CLL generates a broad spectrum of disease-related and treatment-related complications that substantially contribute to morbidity and mortality throughout the disease course. Recognition and prompt management of complications are essential to optimizing patient outcomes. 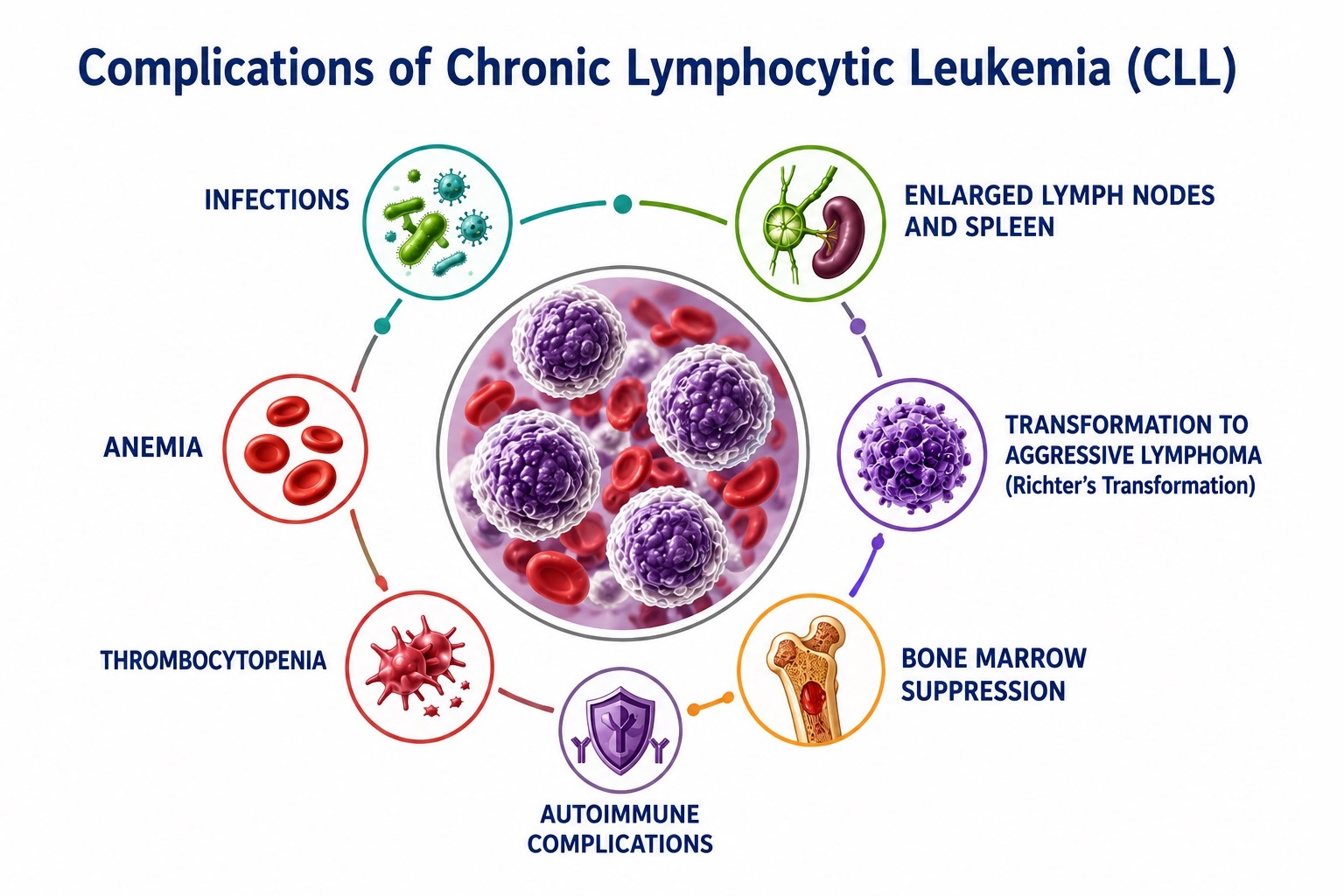
A. Richter Transformation (Richter's Syndrome)
Richter transformation (RT) — the transformation of CLL into a clinically and histologically aggressive lymphoma — occurs in approximately 2–10% of CLL patients, with a lifetime cumulative risk of approximately 10–15%. In approximately 95% of cases, RT manifests as diffuse large B-cell lymphoma (DLBCL), typically arising clonally from the CLL clone (clonal DLBCL) — associated with extremely poor prognosis, with median overall survival of approximately 6–12 months. Rarely (approximately 5%), RT presents as Hodgkin lymphoma transformation or other aggressive histologies.
Clinical Hallmarks: Sudden-onset rapidly enlarging lymph nodes, systemic B symptoms (fever, drenching sweats, marked weight loss), dramatically rising serum LDH, constitutional deterioration. PET-CT shows areas of intense FDG avidity (SUVmax >10). Biopsy of the highest-avidity node is mandatory to confirm transformation histologically. Risk factors include unmutated IGHV, stereotyped IGHV3-21 subset 2, NOTCH1 mutations, TP53 mutations, del(17p), and prior exposure to alkylating agents. Management with R-CHOP or novel clinical trial regimens (anti-PD-1 checkpoint inhibitors, venetoclax combinations) offers limited responses in most patients; allogeneic SCT is considered for those achieving CR.
B. Infectious Complications
- Recurrent bacterial infections (sinopulmonary): The leading cause of morbidity and the most common cause of death in CLL (~50% of mortality). Recurrent pneumonia, bronchitis, sinusitis, and septicemia from encapsulated bacteria (pneumococcus, Haemophilus, Klebsiella) in the setting of hypogammaglobulinemia and neutropenia require aggressive antibiotic management, prophylaxis protocols, and immunoglobulin replacement for patients with IgG <5 g/L and recurrent infections.
- Herpes zoster (shingles): CLL patients have a 10-fold increased risk of varicella zoster virus (VZV) reactivation, including both dermatomal and disseminated presentations. Prophylactic antiviral therapy (acyclovir or valacyclovir) is recommended during and after chemoimmunotherapy and BTK inhibitor therapy.
- Progressive multifocal leukoencephalopathy (PML): Rare but devastating neurological complication from JC virus reactivation, presenting with progressive cognitive decline, motor deficits, speech abnormalities, and visual disturbances. MRI shows characteristic non-enhancing white matter lesions. No proven effective therapy; JC virus PCR from CSF confirms diagnosis. Associated particularly with fludarabine, ofatumumab, and alemtuzumab use.
- COVID-19: CLL patients have markedly elevated COVID-19 mortality risk due to impaired humoral immune responses, poor vaccination efficacy, and the compounding immunosuppression of CLL treatments. Early antiviral therapy (nirmatrelvir-ritonavir — with appropriate drug interactions noted with BTK inhibitors and venetoclax) should be initiated at first symptoms.
C. Autoimmune Cytopenias
- Autoimmune hemolytic anemia (AIHA): Develops in approximately 7–10% of CLL patients; may be spontaneous or precipitated by treatment (notably fludarabine, which should be avoided when DAT-positive). Warm AIHA responds to steroids, rituximab, IVIg, and CLL-directed therapy. Refractory AIHA may require splenectomy or novel agents (sutimlimab for cold agglutinin disease, fostamatinib, rilzabrutinib). AIHA is a source of sudden hemoglobin decline requiring urgent diagnosis to avoid unnecessary blood transfusions and guide appropriate therapy.
- Immune thrombocytopenic purpura (ITP): Autoantibody-mediated platelet destruction; treated with steroids, rituximab, TPO receptor agonists (eltrombopag, romiplostim), and CLL-directed therapy. Distinguishing ITP from marrow failure thrombocytopenia is critical — in ITP, bone marrow shows increased or normal megakaryocytes with adequate erythropoiesis, whereas marrow failure shows hypocellularity with replacement by CLL cells.
- Evans syndrome: The combination of AIHA and ITP occurring simultaneously — a complex and challenging management scenario in CLL requiring combination immunosuppressive and CLL-directed therapy.
D. Second Malignancies
- Skin cancers (SCC, BCC, melanoma): CLL-associated immunosuppression dramatically increases risk — particularly squamous cell carcinoma of the skin, which pursues a more aggressive, metastatic course in CLL patients than in the general population. Annual dermatological surveillance, sun protection, and early biopsy of suspicious skin lesions are recommended for all CLL patients.
- Lung cancer, colorectal cancer, breast cancer: CLL patients have modestly elevated risks of several solid tumors, possibly reflecting shared lifestyle risk factors, treatment-related carcinogenesis (alkylating agents), and immune surveillance failure. Standard cancer screening protocols should be maintained.
- Therapy-related myeloid neoplasm (t-MDS/t-AML): Alkylating agent exposure (chlorambucil, cyclophosphamide, bendamustine) and purine analogue use predispose to therapy-related myelodysplastic syndrome and acute myeloid leukemia as late complications of CLL chemotherapy.
E. Treatment-Related Complications
- Tumor lysis syndrome (venetoclax): Due to BCL-2 inhibition rapidly inducing apoptosis of CLL cells, venetoclax carries a mandatory structured ramp-up dosing schedule (5 weeks from 20 mg to 400 mg) with TLS prophylaxis (allopurinol/rasburicase, aggressive hydration, electrolyte monitoring, hospitalization for high-risk TLS) to prevent hyperkalemia, hyperphosphatemia, hyperuricemia, and renal failure.
- Atrial fibrillation (ibrutinib): Ibrutinib — the first-generation BTK inhibitor — causes clinically significant atrial fibrillation in approximately 10–15% of patients at 3 years, attributed to off-target inhibition of PI3K-delta, c-Src, and EGFR pathways altering cardiac ion channel function. Acalabrutinib and zanubrutinib (second-generation covalent BTK inhibitors) and pirtobrutinib (non-covalent) carry significantly lower AF rates due to improved kinase selectivity. Cardiac monitoring and appropriate anticoagulation management are essential in CLL patients on BTK inhibitors.
- Bleeding (BTK inhibitors): BTK inhibitor-induced impairment of platelet function through collagen receptor signaling inhibition increases bleeding risk — typically minor (bruising, epistaxis, petechiae) but occasionally major (GI bleeding, subdural hematoma). Concomitant antiplatelet or anticoagulant therapy substantially amplifies bleeding risk and requires careful risk-benefit assessment. BTK inhibitors should be held 3–7 days perioperatively.
- Hypertension (ibrutinib): New or worsening hypertension affects approximately 20–30% of ibrutinib-treated patients through inhibition of PI3K-mediated endothelial nitric oxide signaling. Careful blood pressure monitoring and aggressive antihypertensive management are required, as uncontrolled hypertension further amplifies cardiovascular event risk (stroke, MI, AF) in ibrutinib-treated patients.
- Pneumonitis and opportunistic infections (anti-CD20 antibodies, idelalisib): Obinutuzumab and rituximab-based regimens require PCP prophylaxis, antiviral prophylaxis (acyclovir), and hepatitis B reactivation prophylaxis. Idelalisib (PI3K-delta inhibitor) causes a distinctive immune-mediated pneumonitis, colitis, hepatitis, and infectious complications from Pneumocystis and CMV — substantially limiting its clinical use despite initial regulatory approval.
Treatment of Chronic Lymphocytic Leukemia
A fundamental principle of CLL management is that early-stage asymptomatic CLL does not require treatment. The landmark CLL1 and other trials demonstrated no overall survival benefit from early chemotherapy initiation in Binet A or Rai 0 patients — only surveillance ("watch and wait" or "active monitoring") is appropriate until treatment indications are met. Treatment is initiated when patients develop symptomatic disease, progressive cytopenias, or rapidly progressive disease, as defined by iwCLL 2018 treatment criteria.
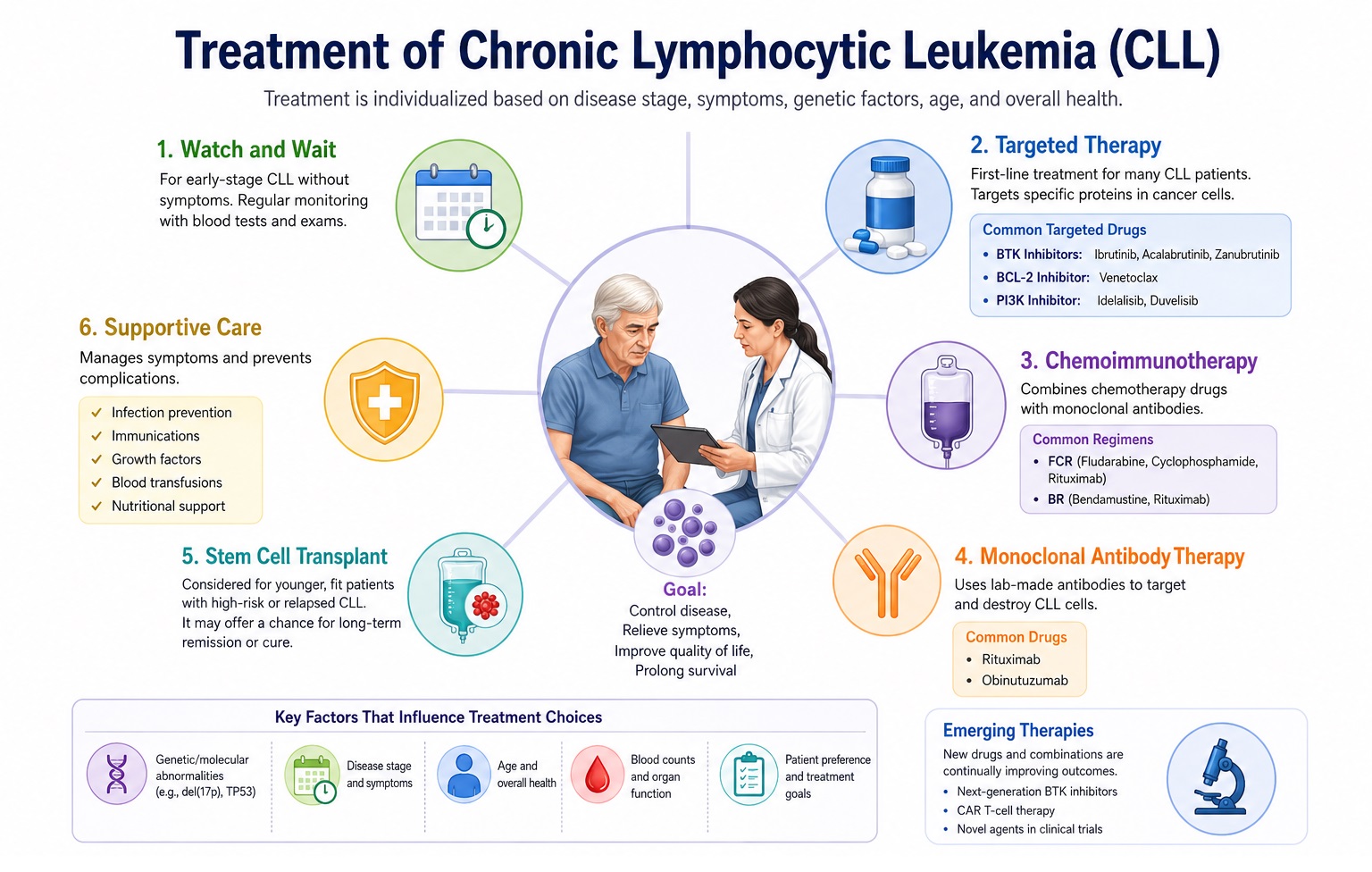
A. Frontline Treatment — Fit Patients (CIRS ≤6, CrCl ≥70 mL/min)
Ibrutinib (420 mg orally once daily, continuous until progression or toxicity) was the first BTK inhibitor to demonstrate superior progression-free and overall survival versus chemoimmunotherapy in both previously untreated and relapsed/refractory CLL patients across multiple landmark randomized trials (RESONATE, RESONATE-2, ALLIANCE, ECOG-ACRIN E1912, iLLUMINATE). Ibrutinib is effective even in del(17p)/TP53-mutated CLL — historically untreatable with chemotherapy — producing durable responses without complete remissions in most patients. However, ibrutinib's cardiac toxicity profile (AF, hypertension, bleeding) has prompted evaluation of second-generation agents.
Acalabrutinib (100 mg twice daily) and zanubrutinib (160 mg twice daily or 320 mg once daily) are second-generation, more selective covalent BTK inhibitors with significantly improved cardiac tolerability (AF rate ~3–5% vs ~12–15% with ibrutinib). ALPINE trial demonstrated superior progression-free survival and lower AF with zanubrutinib versus ibrutinib in relapsed/refractory CLL, establishing zanubrutinib as the preferred BTK inhibitor at many centers.
The CLL14 trial established fixed-duration venetoclax (BCL-2 inhibitor) combined with obinutuzumab (anti-CD20 antibody) for 12 months as a frontline standard of care for previously untreated fit and unfit CLL, demonstrating superior progression-free survival compared to chlorambucil-obinutuzumab at 6-year follow-up (median PFS not reached vs 27 months for control). Critically, this regimen achieves high rates of undetectable MRD (uMRD) in peripheral blood (~75%) and bone marrow (~57%), associated with prolonged remission duration. The fixed-duration nature — 12 months vs continuous BTK inhibitor therapy — and the resulting treatment-free interval offer major patient quality-of-life advantages. VenG is particularly preferred for del(11q) and mutated IGHV CLL.
Chemoimmunotherapy with FCR — combining fludarabine (25 mg/m² D1–3), cyclophosphamide (250 mg/m² D1–3), and rituximab (375–500 mg/m²) for 6 cycles — historically represented the standard of care for young, fit CLL patients. The CLL8 trial and long-term follow-up demonstrated that FCR achieves complete remission in approximately 44% of patients, with a landmark plateau in progression-free survival curves at 10+ years in patients with mutated IGHV (approximately 40% remain treatment-free at 12 years). FCR is now generally reserved for young (<65 years), biologically fit patients with mutated IGHV and favorable cytogenetics (no del(17p), del(11q), or TP53 mutation), given its substantial and durable remissions in this genomically favorable subgroup. FCR is contraindicated in TP53-mutated/del(17p) CLL and should be used with caution in del(11q).
The GLOW and CAPTIVATE trials evaluated the fixed-duration combination of ibrutinib plus venetoclax for 12–15 months, demonstrating high uMRD rates (>55% in bone marrow) and durable PFS superior to conventional chemoimmunotherapy in frontline CLL including high-risk genomic subgroups. This doublet-targeted approach, combining BCR pathway suppression (BTK inhibition) with anti-apoptotic blockade (BCL-2 inhibition), represents the emerging paradigm for "chemotherapy-free, fixed-duration" deepest possible remission — ongoing trials exploring MRD-guided treatment cessation.
B. Frontline Treatment — Unfit / Elderly Patients (CIRS >6 or CrCl <70 mL/min)
- Venetoclax + Obinutuzumab (VenG): As described above — CLL14 demonstrated superiority even in the unfit patient population over chlorambucil-obinutuzumab; fixed-duration 12 months with compelling efficacy and manageable toxicity in elderly and comorbid patients. Preferred by many guidelines as frontline regimen for unfit patients with del(17p)/TP53 mutation.
- BTK inhibitors (acalabrutinib, zanubrutinib, ibrutinib): Well tolerated in elderly patients; continuous oral therapy; no myelosuppression; appropriate for patients with renal impairment (no dose adjustment required for zanubrutinib in renal dysfunction) and multiple comorbidities, provided cardiac status and bleeding risk are managed.
- Chlorambucil + anti-CD20 antibody (obinutuzumab or rituximab): For frail patients unable to tolerate BTK inhibitors or venetoclax — chlorambucil-obinutuzumab (CLL11 trial; superior to chlorambucil alone) or chlorambucil-rituximab provides modest but clinically meaningful responses in the most frail CLL populations. Role substantially diminished in modern era.
C. Relapsed/Refractory CLL Treatment
- BTK inhibitor → venetoclax-based regimen switch: Patients relapsing on continuous BTK inhibitor therapy typically transition to venetoclax-based treatment (venetoclax + rituximab — MURANO trial; or venetoclax + obinutuzumab). MURANO demonstrated superior PFS for venetoclax-rituximab (24 months fixed-duration) vs bendamustine-rituximab in relapsed/refractory CLL with high uMRD rates and significant OS benefit.
- Pirtobrutinib (non-covalent BTK inhibitor): The first non-covalent (reversible) BTK inhibitor, pirtobrutinib binds BTK in a manner that retains activity against the C481S BTK resistance mutation — the most common mechanism of acquired BTK inhibitor resistance. Approved for CLL/SLL after at least 2 prior lines of therapy including a covalent BTK inhibitor. BRUIN trial demonstrated 73% overall response rate in BTK inhibitor-pretreated patients, including those with C481S mutation. Represents a major advance for patients failing covalent BTK inhibitors.
- Lisocabtagene maraleucel (liso-cel) — CAR-T cell therapy: The first CAR-T cell therapy FDA-approved for CLL (TRANSCEND-CLL-004 trial), indicated for adult patients with relapsed/refractory CLL or SLL after at least two prior lines of therapy, including a BTK inhibitor and venetoclax. Achieved 43% complete remission rate and 20% uMRD-negative complete remission in a highly pretreated patient population. Represents a potential new curative-intent approach for multiply relapsed CLL.
- Allogeneic hematopoietic stem cell transplantation (Allo-SCT): The only proven curative therapy for CLL, reserved for young (<65 years), fit patients with del(17p)/TP53-mutated disease, Richter transformation achieving remission with salvage therapy, or patients with multiply relapsed disease after targeted therapies. Reduced-intensity conditioning (RIC) allo-SCT minimizes transplant-related mortality in older/comorbid patients while preserving the graft-versus-leukemia effect. Transplant-related mortality is approximately 10–20% at experienced centers.
- PI3K-delta inhibitor (idelalisib + rituximab): Second-line option for relapsed/refractory CLL — superior PFS vs rituximab alone (STUDY 116) but substantially limited by immune-mediated toxicities (colitis, hepatotoxicity, pneumonitis, infections) requiring close monitoring and mandatory PCP/antifungal/antiviral prophylaxis.
D. Special Treatment Situations
- Autoimmune cytopenias in CLL: AIHA and ITP — treated with corticosteroids (prednisolone 1 mg/kg/day) as first-line; rituximab for steroid-refractory cases; IVIg for acute bleeding in ITP; CLL-directed therapy (BTK inhibitor preferred as it does not worsen autoimmune cytopenias) to control underlying disease. Fludarabine must be avoided in DAT-positive AIHA.
- Richter transformation to DLBCL: Treatment with R-CHOP-based chemoimmunotherapy; partial responses are common but durable CR is uncommon in clonal transformation. Checkpoint inhibitors (pembrolizumab, nivolumab) have shown activity in RT. Allogeneic SCT for those achieving CR. Novel trials with CAR-T therapy are ongoing.
- Supportive care: Immunoglobulin replacement therapy (IVIG or subcutaneous Ig — SCIg) for patients with IgG <5 g/L and recurrent bacterial infections; granulocyte colony-stimulating factor (G-CSF) for treatment-related febrile neutropenia; antibiotic/antifungal/antiviral prophylaxis during intensive therapy; pneumococcal, influenza, Hib, and herpes zoster vaccines (recombinant/inactivated — not live) for all CLL patients.
Prevention and Risk Reduction in CLL
Unlike many solid tumors and acute leukemias, CLL lacks well-established modifiable environmental risk factors amenable to large-scale primary prevention programs — as the dominant predisposing factors are genetic (inherited susceptibility loci, family history). However, evidence-based strategies can reduce risk in susceptible populations, facilitate early detection, prevent disease complications, and optimize outcomes in established disease. 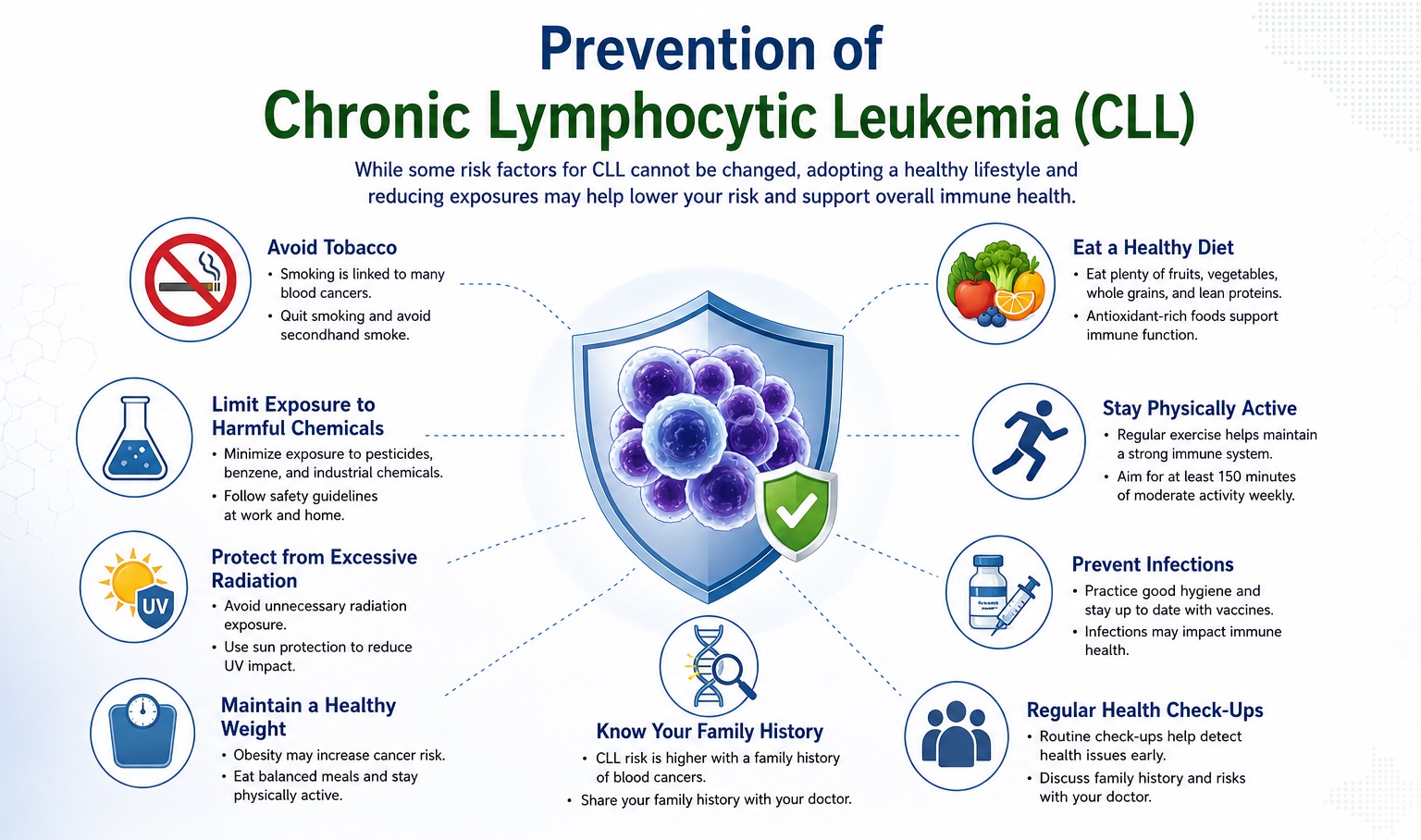
A. Primary Prevention — Reducing CLL Incidence
- Avoidance of occupational and environmental chemical exposures: Minimizing exposure to established CLL-linked carcinogens — particularly organochlorine pesticides, phenoxyacid herbicides, and benzene derivatives — through occupational safety regulations, personal protective equipment compliance, safe agricultural practices, and regulatory control of leukemogenic chemical exposures. Veterans with documented Agent Orange exposure should receive surveillance for MBL and early CLL, given the VA-recognized causal relationship.
- Hepatitis C virus (HCV) prevention and treatment: Universal HCV screening and treatment with highly effective direct-acting antivirals eliminates chronic antigenic B-cell stimulation linked to CLL pathogenesis. HCV eradication not only prevents cirrhosis and hepatocellular carcinoma but may reduce CLL risk and, in rare cases, benefit established CLL through immune reconstitution.
- Tobacco cessation: While the CLL-smoking association is less robust than for other hematological malignancies, the broad cardiovascular, pulmonary, and second-malignancy risk reduction of smoking cessation benefits CLL patients — who already face elevated second malignancy risk from disease-related immunosuppression.
- Regular physical activity and healthy body weight: Moderate regular physical exercise reduces systemic inflammation, improves immune function, and may modestly reduce lymphoma/leukemia risk. Physical activity programs in CLL patients improve fatigue, quality of life, and cardiorespiratory fitness — counteracting the deconditioning of chronic disease.
- Ultraviolet radiation protection: Given the dramatically elevated risk of skin cancers — particularly squamous cell carcinoma — in CLL patients, rigorous sun protection (SPF 50+ sunscreen, protective clothing, limiting peak sun exposure) and annual dermatological examination are critical preventive measures for established CLL patients.
B. Familial CLL Surveillance and Genetic Counseling
- First-degree relative monitoring programs: Given the 7–9-fold elevated CLL risk and 13–18% MBL prevalence in first-degree relatives of CLL patients, structured clinical genetics programs offering CLL family registration, annual CBC monitoring, and flow cytometric MBL screening for willing family members enable early detection of MBL and pre-symptomatic CLL — maximizing the "watch and wait" period and ensuring timely treatment initiation when indications arise.
- Genetic counseling: Recommended for CLL patients with strong family histories, early onset (<50 years), or multiple affected relatives. Genetic counselors can provide risk assessment, coordinate germline sequencing (panel testing for relevant susceptibility loci in research settings), and counsel family members on risk implications and surveillance options.
- MBL surveillance protocols: Individuals identified with high-count MBL (≥0.5 × 10⁹/L) — progressing to CLL at ~1–2% per year — benefit from annual CBC and clinical assessment at minimum, with flow cytometry monitoring and clinical vigilance for new lymphadenopathy, organomegaly, or cytopenias signaling CLL development.
C. Infection Prevention in Established CLL
- Vaccination programs: All CLL patients should receive annual influenza vaccination (inactivated), pneumococcal vaccines (PCV20 or PCV13 followed by PPSV23), Haemophilus influenzae type b (Hib) conjugate vaccine, and recombinant zoster vaccine (Shingrix — indicated despite immunodeficiency; live varicella vaccine is contraindicated). COVID-19 vaccination (mRNA vaccines preferred — 3-dose primary series + boosters) is recommended, recognizing limited but potentially meaningful immunogenicity. Hepatitis B vaccination for seronegative patients undergoing anti-CD20 antibody therapy; hepatitis B reactivation prophylaxis with entecavir for all HBsAg+ or anti-HBc+ patients receiving rituximab/obinutuzumab.
- Immunoglobulin replacement therapy (IRT): Indicated for patients with IgG <5 g/L and ≥2 significant bacterial infections per year despite vaccination and antibiotic prophylaxis. Monthly IV immunoglobulin (400–600 mg/kg) or biweekly subcutaneous immunoglobulin administration reduces the frequency of serious bacterial infections and hospitalizations for infection in hypogammaglobulinemic CLL patients by approximately 50–60%.
- Antimicrobial prophylaxis during treatment: PCP prophylaxis (trimethoprim-sulfamethoxazole 960 mg three times weekly, or dapsone/atovaquone for sulfonamide-intolerant patients) mandatory during and for 6–12 months after purine analogue-containing, obinutuzumab-based, or idelalisib-based therapy. Antiviral prophylaxis with acyclovir or valacyclovir for HSV and VZV reactivation during intensive therapy. Antifungal prophylaxis in prolonged neutropenia.
- Hepatitis B reactivation prevention: All CLL patients receiving anti-CD20 antibody therapy (rituximab, obinutuzumab) must be screened for hepatitis B surface antigen (HBsAg) and core antibody (anti-HBc). HBsAg-positive or anti-HBc-positive patients require prophylactic antiviral therapy with entecavir or tenofovir, continued for at least 18 months after completion of anti-CD20 therapy, due to the risk of potentially fatal hepatitis B reactivation.
D. Prevention of CLL Complications and Treatment Toxicities
- Richter transformation surveillance: Clinical vigilance for clinical features suggesting RT (sudden lymph node enlargement, B symptoms, sharply rising LDH, rapid deterioration) with prompt PET-CT and node biopsy is essential. Earlier detection of RT maximizes the probability of achieving CR with salvage therapy and consideration of allogeneic SCT.
- Tumor lysis syndrome prevention with venetoclax: Mandatory structured venetoclax dose ramp-up (20→50→100→200→400 mg over 5 weeks), baseline TLS risk assessment per clinical, laboratory, and radiographic parameters, allopurinol or rasburicase prophylaxis, aggressive intravenous hydration, electrolyte monitoring, and inpatient observation for high-risk TLS patients are non-negotiable components of venetoclax safety management.
- Atrial fibrillation and cardiovascular monitoring on BTK inhibitors: Baseline and periodic cardiovascular assessment (ECG, blood pressure, cardiac history review) for all patients on BTK inhibitors. Prompt anticoagulation decision-making (balancing CLL-related bleeding risk with AF-related stroke risk) when ibrutinib-associated AF develops; consideration of BTK inhibitor switch to acalabrutinib or zanubrutinib for ibrutinib-intolerant patients.
- Skin cancer prevention and surveillance: Annual dermatological examinations with total body skin survey; rigorous sun protection measures; early biopsy and definitive treatment of skin lesions — particularly SCC in situ and invasive SCC, which may pursue aggressive, metastatic courses in immunosuppressed CLL patients.
- Psychosocial support and quality of life programs: CLL-specific support groups, psychological counseling, mindfulness-based stress reduction, structured exercise programs, and patient education are validated components of comprehensive CLL care that improve quality of life, treatment adherence, and overall well-being.
Common FAQs on Chronic Lymphocytic Leukemia
Bibliography on Chronic Lymphocytic Leukemia
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.
- Wierda WG, Byrd JC, O'Brien SM, et al. NCCN Clinical Practice Guidelines in Oncology: Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Version 3.2024. National Comprehensive Cancer Network.
- Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2019;381(5):432–443. [ECOG-ACRIN E1912]
- Al-Sawaf O, Zhang C, Tandon M, et al. Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): 6-year follow-up of an open-label, randomised, phase 3 trial. Lancet Oncology. 2023;24(2):212–227.
- Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax–Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2018;378(12):1107–1120. [MURANO Trial]
- Tam CS, Giannopoulos K, Robak T, et al. GLOW: Ibrutinib plus venetoclax for first-line treatment of chronic lymphocytic leukemia. Journal of Clinical Oncology. 2022;40(25):2969–2978.
- Brown JR, Eichhorst B, Hillmen P, et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2023;388(4):319–332. [ALPINE Trial]
- Byrd JC, Hillmen P, Ghia P, et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. Journal of Clinical Oncology. 2021;39(31):3441–3452. [ELEVATE-RR]
- Tam CS, Allan JN, Siddiqi T, et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood. 2022;139(22):3278–3289.
- Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncology. 2016;17(6):768–778. [M13-982 Trial]
- Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2015;373(25):2425–2437. [RESONATE-2 Trial]
- Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772.
- International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI). Lancet Oncology. 2016;17(6):779–790.
- Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. New England Journal of Medicine. 2019;380(23):2225–2236. [CLL14 Trial]
- Williams Hematology, 10th ed. Editors: Kaushansky K, Prchal JT, Burns LJ, et al. McGraw-Hill Education. Chapter: Chronic Lymphocytic Leukemia.