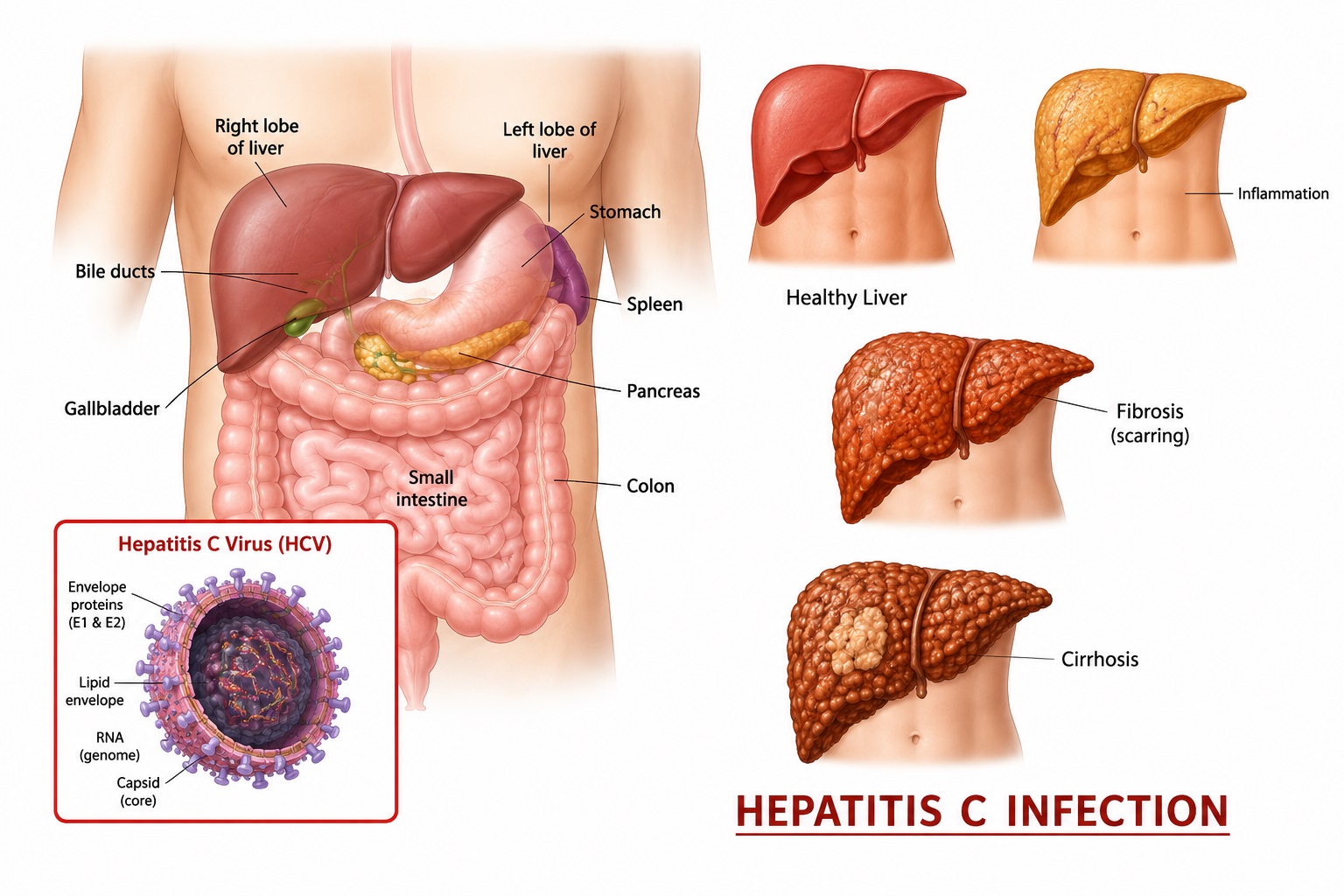


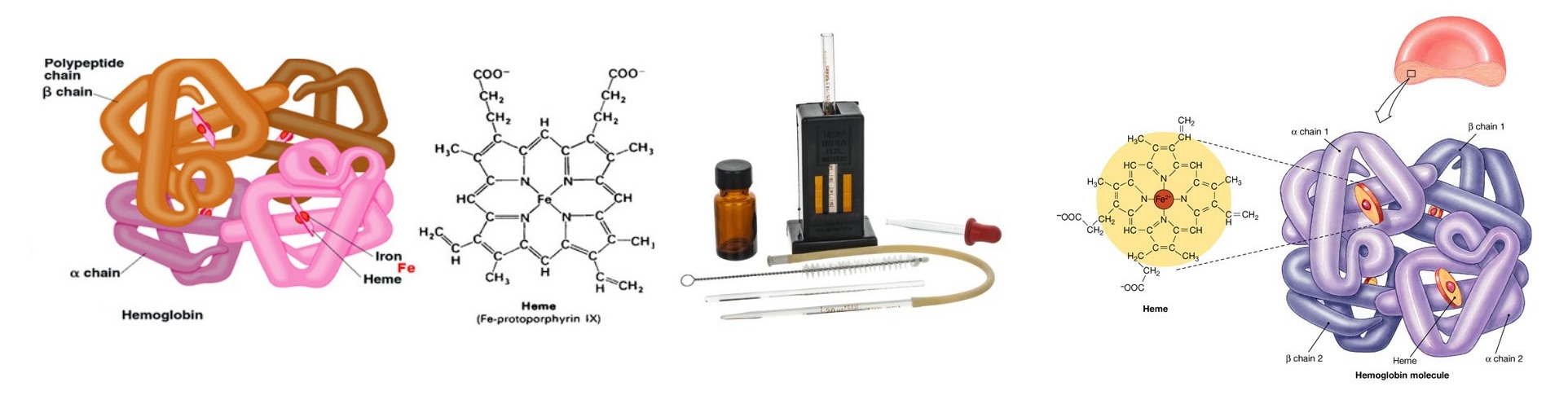
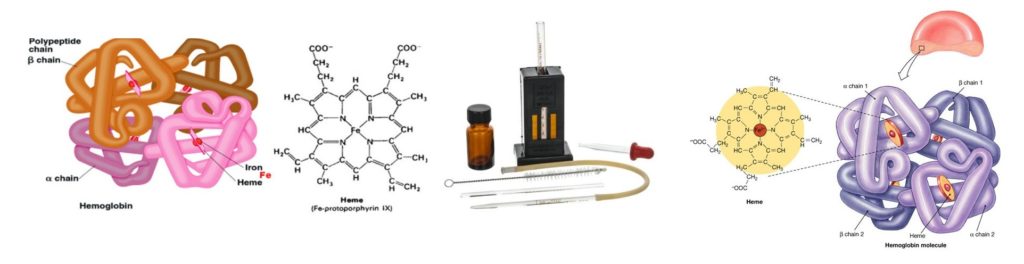
Hemoglobin Estimation : Significance, Reference Range, Procedure…..
Hemoglobin is a conjugated protein present inside the erythrocytes. Hemoglobin consist of a prosthetic group named heam, which is combined with protein called globin (Hemoglobin = Heam + Globin). Other heam containing proteins in the body are Myoglobin, Cytochrome-C etc. Heam carries oxygen (O2) from the lungs to the tissue cells and carbon dioxide (CO2), the gaseous waste product from the cells to the lungs. After the normal life span of RBC (over 120 days), the red cells are destroyed by the reticuloendothelial cells (specially in the spleen) and the components of the hemoglobin undergo metabolic degeneration.
Several methods are available for the estimation of hemoglobin in the blood. These are:
- Acid heamatin method (Sahli’s method)
- Cyanmethemoglobin method.
- Alkaliheamatin method.
- Haldane’s Carboxyhemoglobin method.
- Oxyhemoglobin method.
Former two methods are commonly used for determination of hemoglobin concentration.
Clinical Significance :
A decreased in hemoglobin concentration below normal range in an indication of anaemia. An increased in hemoglobin concentration occurs in haemo-concentration due to loss of body fluid in sever diarrhea and vomiting. High value also observed in congenital heart disease (due to reduce O2 supply) in emphysema and also in Polycythemia.
Hemoglobin concentration drops during pregnancy due to haemodilution (a proportionately greater increase in plasma volume as compared to the increase in the red cell mass). Several abnormal forms of hemoglobin (variants) have been identified in the human body but only a few are common and clinically significant.
Common hemoglobin variants :
- Hemoglobin S: This is the primary hemoglobin present in the human body with Sickle Cell Disease (also known as Sickle Cell Anemia). Approximately 0.35% of African American babies are born with sickle cell disease and about 100,000 Americans live with the disorder according to the Centers for Disease Control and Prevention. Sickle cell anaemia patient's hemoglobin have two abnormal beta chains and two normal alpha chains.
Presence of Hemoglobin S causes red blood cell to deform and assume a sickle shape when exposed to decreased amounts of oxygen; when someone exercises or has pulmonary infection. Sickled red blood cells are rigid and can block small blood vessels, causing pain, impaired circulation and decreased oxygen delivery as well as shortened red cell survival. A single beta (βS) copy (sickle cell trait) typically does not cause significant symptoms unless it is combined with another hemoglobin mutation, such as that causing HbC or beta thalassemia.
- Hemoglobin C: About 2-3% of African Americans in the United States are heterozygotes for Hemoglobin C (Hemoglobin C trait) and are often asymptomatic. Hemoglobin C disease is rare about 0.02% of African Americans population and relatively mild. It usually causes a minor amount of haemolytic anaemia and a mild to moderate enlargement of the spleen.
- Hemoglobin E: Hemoglobin E is one of the most common beta chain hemoglobin variants in the world. It is very prevalent in Southeast Asia, especially in Cambodia, Laos, and Thailand, and in individuals of Southeast Asian descent. People who are homozygous for HbE (have two copies of βE) generally have a mild hemolytic anemia, microcytic red blood cells, and a mild enlargement of the spleen. A single copy of the hemoglobin E gene does not cause symptoms unless it is combined with another mutation, such as the one for beta thalassemia trait.
Less common hemoglobin variants :
There are many other variants. Some are silent–causing no signs or symptoms–while others affect the functionality and/or stability of the hemoglobin molecule. Examples of other variants include: Hemoglobin D, Hemoglobin G, Hemoglobin J, Hemoglobin M, and Hemoglobin Constant Spring caused by a mutation in the alpha-globin gene that results in an abnormally long alpha (α) chain and an unstable hemoglobin molecule. Additional examples include:
- Hemoglobin F (HbF): HbF is the primary hemoglobin produced by the fetus and it's transport oxygen efficiently in a low oxygen environment. Production of HbF decreases sharply after birth and reaches adult levels by 1-2 years of age. HbF may be elevated in several congenital disorders. Levels can be normal to significantly increased in beta thalassemia and are frequently increased in individuals with sickle cell anemia and in sickle cell-beta thalassemia. Individuals with sickle cell disease and increased HbF often have a milder disease, as the HbF inhibits sickling of the red cells.
HbF levels are also increased in a rare condition called Hereditary Persistence of Fetal Hemoglobin (HPFH). This is a group of inherited disorders in which HbF levels are increased without the any signs & symptoms of thalassemia. Different ethnic groups have different mutations causing HPFH. HbF can also be increased in some acquired conditions involving impaired red blood cell production. Some leukemias and other myeloproliferative neoplasms are also associated with mild elevation in HbF. - Hemoglobin H: HbH is an abnormal hemoglobin that occurs in some cases of alpha thalassemia. It is composed of four beta (β) globin chains and is produced due to a severe shortage of alpha (α) chains. Although each of the beta (β) globin chains is normal, the tetramer of 4 beta chains does not function normally. It has an increased affinity for oxygen, holding onto it instead of releasing it to the tissues and cells. Hemoglobin H is also associated with significant breakdown of red blood cells as it is unstable and tends to form solid structures within red blood cells. Serious medical problems are not common in people with hemoglobin H disease, though they often have anemia.
- Hemoglobin Barts: Hb Barts develops in fetuses with alpha thalassemia. It is formed of four gamma (γ) protein chains when there is a shortage of alpha chains, in a manner similar to the formation of Hemoglobin H. If a small amount of Hb Barts is detected, it usually disappears shortly after birth due to dwindling gamma chain production. These children have one or two alpha gene deletions and are silent carriers or have the alpha thalassemia trait. If a child has a large amount of Hb Barts, he/she usually has hemoglobin H disease and a three-gene deletion. Fetuses with four-gene deletions have hydrops fetalis and usually do not survive without blood transfusions and bone marrow transplants.
A person can also inherit two different abnormal genes, one from each parent. This is known as being compound heterozygous or doubly heterozygous. Several different clinically significant combinations are listed below.....
- Hemoglobin SC disease: Inheritance of one beta S gene and one beta C gene results in Hemoglobin SC disease. These individuals have a mild hemolytic anemia and moderate enlargement of the spleen. Persons with HbSC disease may develop the same vaso-occulsive (blood vessel-blocking) complications as seen in sickle cell anemia but most cases are less severe.
- Sickle Cell-Hemoglobin D disease: Individuals with sickle cell-HbD disease have inherited one copy of hemoglobin S and one of hemoglobin D-Los Angeles (or D-Punjab) genes. These people may have occasional sickle crises and moderate hemolytic anemia.
- Hemoglobin E-beta thalassemia: Individuals who are doubly heterozygous for hemoglobin E and beta thalassemia have an anemia that can vary in severity from mild (or asymptomatic) to severe, dependent on the beta thalassemia mutation(s) present.
- Hemoglobin S-beta thalassemia: Sickle cell-beta thalassemia varies in severity, depending on the beta thalassemia mutation inherited. Some mutations result in decreased beta globin production (beta+) while others completely eliminate it (beta0). Sickle cell-beta+ thalassemia tends to be less severe than sickle cell-beta0 thalassemia. People with sickle cell beta0 thalassemia tend to have more irreversibly sickled cells, more frequent vaso-occlusive problems and more severe anemia than those with sickle cell-beta+thalassemia. It is often difficult to distinguish between sickle cell disease and sickle cell-beta0 thalassemia.
Normal values :
- Male : 13 – 18 gm/dl
- Female : 12 – 16.5 gm/dl
- Children (up to 1 year) : 11 – 13 gm/dl
- Children (10-12 years) : 11.5 – 14.5 gm/dl
- Infants (New born) : 13.5 – 19.5 gm/dl
Determination of Hemoglobin by Cyanmethemoglobin metnod
Principle :
When anticoagulated blood is mixed with Drabkin’s reagent containing potassium cyanide and potassium fericyanide. Hemoglobin reacts with potassium fericyanide to form methemoglobin which is converted to stable Cyanmethemoglobin by the potassium cyanide. The intensity of the colour is directly proportional to the concentration of the concentration of hemoglobin present in the specimen. The intensity of the colour is measured in the colorimeter at 540 nm (green filter).
Specimen :
Free flowing capillary blood or anticoagulated venous blood (EDTA or Double oxalate). The specimen need not be fasting sample.
Requirements :
- Drabkin’s reagent :
- Potassium cyanide : 100 mg
- Potassium fericyanide : 400 mg
- Potassium dihydrogen phosphate : 280 mg
- Nonidet : 1.0 ml
- Distilled water : 1000 ml
- Cyanmethemoglobin standard – 15 gm/dl
- Micropipette with micro-tips.
- Test tube.
- 5 ml pipette.

Procedure :
- Pipettes in the tubes labeled as following :
| Reagent | Test (T) | Blank (B) |
| Drabkin’s reagent, ml
Blood, ml (well mixed) |
5.0
0.02 |
5.0
– |
- Mixed the contains in the tube labeled as ‘Test’ thoroughly and wait for 5 minutes.
- Read the absorbance of the ‘Test’ by setting ‘Blank’ to 100% T at 540 nm (green filter) in the colorimeter.
- Read the absorbance of the standard (15 gm/dl) by pipettes it directly in a cuvette.
- Calculation :

Preparation of Standard Graph :
A standard graph for determination of hemoglobin can be prepared as follows :
Requirements :
- Drabkin’s reagent
- Cyanmethemoglobin standard – 15 gm/dl.
Procedure :
- Pipettes in the tubes labeled as following :
| Reagent | Std. 5 | Std. 10 | Std. 15 | Blank |
| Drabkin’s reagent, ml | 3.34 | 1.67 | 0.00 | 5.0 |
| Hemoglobin Standard, ml | 1.66 | 3.33 | 5.0 | 0.00 |
- Mixed well and wait for 3 minutes and read the absorbance of colour of these Standards by setting Blank to 100% T at 540 nm (green filter) in the colorimeter.
- Prepared a graph by plotting Absorbance of standard on ‘Y’ axis and Concentration of hemoglobin Standard that is 5.0 gm/dl, 10.0 gm/dl and 15.0 gm/dl on ‘X’ axis.
- A standard line passing through origin indicates agreement with “Beer’s law”.
- This Graph can be used as a Standard graph for hemoglobin concentration determination.
Determination of Hemoglobin by Acidhematin method
Principle :
When blood is added with 0.1(N) HCl, hemoglobin is converted to brown coloured acid-haematin. The resulting colour after dilution with distilled water is compared with standard brown glass reference blocks of a Sahli’s hemoglobinometer.
Requirements :
- Sahli’s hemoglobinometer :
- Glass comparator.
- Sahli’s glass tube.
- Hb-pipette (0.02 ml)
- 1(N)HCl acid.
- Distilled water
- Pasteur pipette.
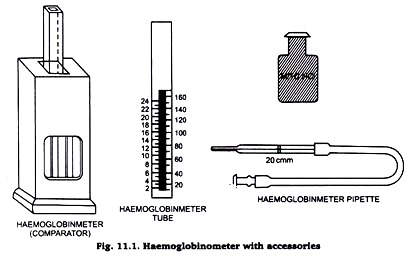
Procedure :
- Placed 0.1(N)HCl acid in the Sahli’s tube upto the lowest mark 20% by using a Pasteur pipette
- Pipette 0.02 ml of blood in a Hb-pipette and added with the 0.1(N)HCl acid present in the Sahli’s tube. Mixed well and wait for 10 minutes.
- Diluted the solution with distilled water by adding few drops at a time carefully and diluted the solution, until the colour matches with the glass comparator present in the haemometer.
- The colour matching should be done only against natural day light. The level of the fluid is noted at its lower meniscus and the reading corresponding to this level on the scale is recorded in gm/dl.
Additional Information :
- Methemoglobin, Carboxyhemoglobin and sulfhemoglobin are not converted to acid haematin by reacts with 0.1(N)HCl acid.
- This method is useful for places where photometers are not available.
- It can give an error upto 1.0 gm/dl.