

Chronic Myeloid Leukemia (CML) – Sign and Symptoms, Risk Factors, Diagnosis, Complications, Treatment and Prevention
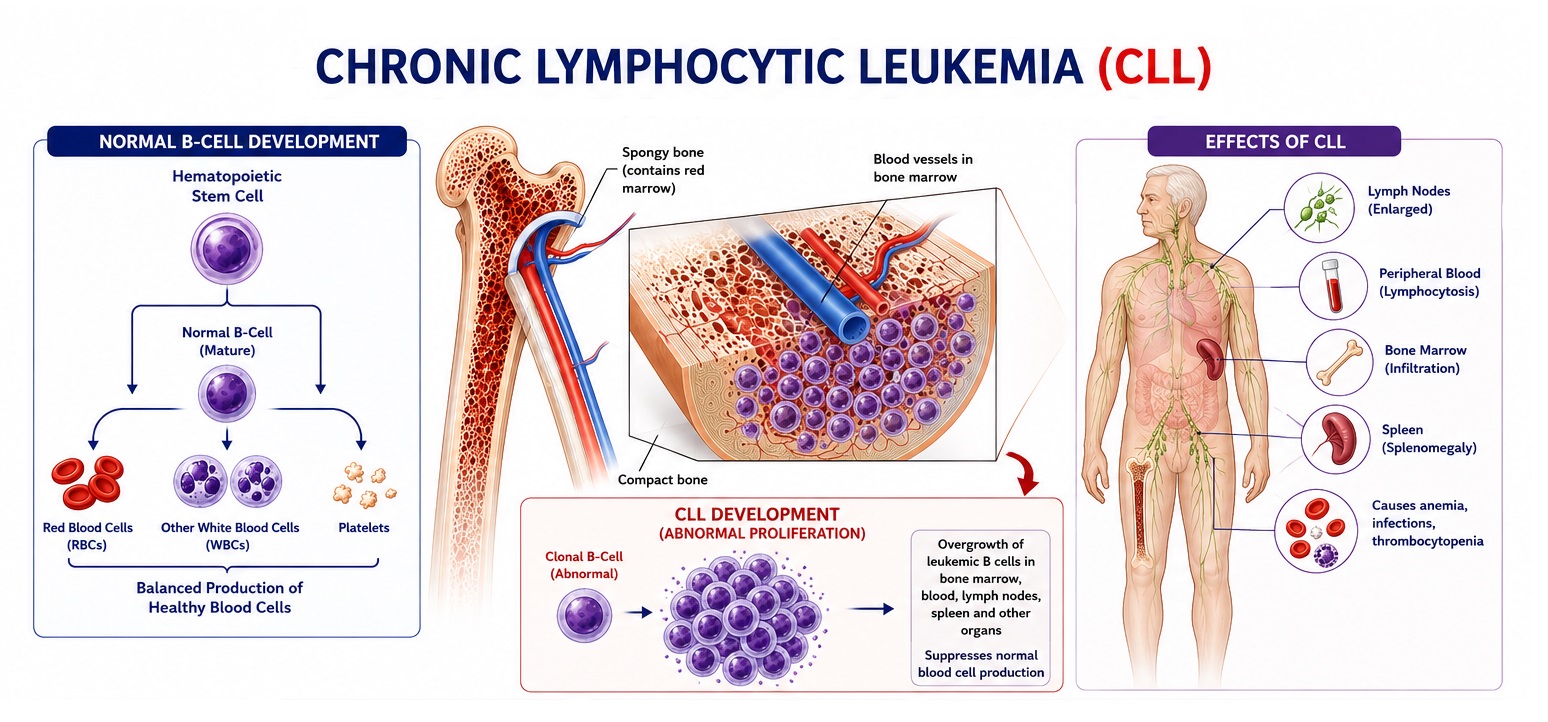
Chronic Myeloid Leukemia (CML) is a clonal myeloproliferative neoplasm arising from a pluripotent hematopoietic stem cell and defined at the molecular level by the BCR-ABL1 fusion gene. This oncogene is created by the reciprocal chromosomal translocation t(9;22)(q34;q11.2), which produces the characteristic Philadelphia chromosome (Ph chromosome) — a shortened chromosome 22 first identified in 1960 by Nowell and Hungerford. The BCR-ABL1 protein is a constitutively active non-receptor tyrosine kinase that drives uncontrolled proliferation, impaired apoptosis, and genomic instability of myeloid progenitor cells, resulting in the massive accumulation of mature and maturing granulocytes in the peripheral blood, bone marrow, and extramedullary sites such as the spleen and liver. 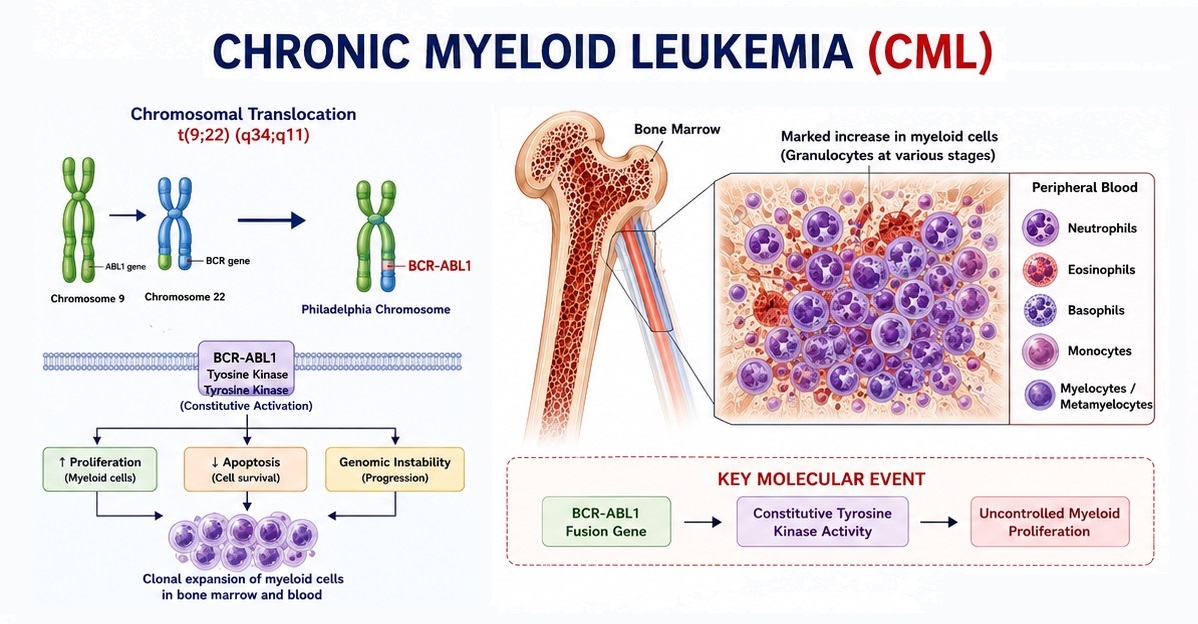
CML accounts for approximately 15–20% of all leukemias in adults, with a global incidence of 1–2 cases per 100,000 population annually. The median age at diagnosis is 50–60 years, though it can occur at any age including in children and adolescents. There is a slight male predominance (male-to-female ratio ≈ 1.3:1). The natural history of CML is classically triphasic: an indolent Chronic Phase (CP) lasting 3–6 years, an Accelerated Phase (AP) with increasing disease burden over several months, and a terminal Blast Phase (BP, or blast crisis) that clinically resembles acute leukemia and carries a dismal prognosis without intervention.
The management of CML was transformed by the introduction of imatinib mesylate (Gleevec/Glivec), the first BCR-ABL1-specific tyrosine kinase inhibitor (TKI), in 2001. Its remarkable efficacy led to a paradigm shift in oncology — the targeted molecular therapy model. Subsequent generations of TKIs (dasatinib, nilotinib, bosutinib, and ponatinib) further improved response rates and overcame resistance. Today, the majority of CML patients in chronic phase who receive TKI therapy achieve durable remissions, with 10-year survival rates exceeding 85–90%, approaching that of the general population. A subset of deeply responding patients may even achieve Treatment-Free Remission (TFR), the modern equivalent of a functional cure. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains an option for patients with TKI resistance or advanced-phase disease.
Sign and Symptoms of Chronic Myeloid Leukemia (CML)
The clinical manifestations of CML vary considerably depending on the phase of disease at presentation, the degree of leukocytosis, spleen size, and whether constitutional or systemic features are present. Approximately 40–50% of patients in the chronic phase are asymptomatic at diagnosis, with the disease discovered incidentally on a routine complete blood count (CBC). As the disease progresses from chronic to accelerated and blast phase, symptoms become progressively more severe, reflecting increasing tumor burden, bone marrow failure, and organ infiltration.
Chronic Phase (CP) : Often asymptomatic or mildly symptomatic. Dominated by massive leukocytosis, splenomegaly, and constitutional features. Accounts for ~85% of newly diagnosed patients.
Accelerated Phase (AP) : Worsening constitutional symptoms, increasing splenomegaly, cytopenias, new cytogenetic abnormalities, and increasing blast percentage (10–19%).
Blast Phase (BP) : Acute leukemia-like picture: ≥20% blasts in blood/marrow, severe systemic symptoms, organomegaly, bone pain, and risk of leukostasis emergency.
1. Chronic Phase Manifestations
A. Constitutional Symptoms
- Fatigue and generalized weakness: The most commonly reported symptom, resulting from anemia secondary to replacement of erythroid progenitors by expanding myeloid mass in the bone marrow, and from increased metabolic demands of proliferating leukemic cells. Fatigue may progress insidiously over weeks to months and significantly impairs daily functioning and quality of life.
- Unintentional weight loss: Reflects the high catabolic state driven by rapid cell turnover and elevated cytokine levels (IL-6, TNF-α). Weight loss exceeding 10% of body weight over 6 months indicates aggressive disease and may herald phase transition.
- Night sweats: Profuse nocturnal diaphoresis resulting from cytokine-mediated thermoregulatory disturbances and hypermetabolism of proliferating myeloid cells. Night sweats may disturb sleep and contribute to general debility.
- Low-grade fever: Occurs due to pyrogen release from cytokine cascades rather than infection in early stages. However, fever in later stages should always prompt evaluation for secondary infections.
- Generalized malaise and reduced exercise tolerance: Reflecting the combined effects of anemia, cytokine activity, and increased metabolic demands of the expanded leukemic clone.
B. Splenomegaly-Related Symptoms
Splenomegaly is the most characteristic physical finding in CML, present in approximately 50–70% of patients at diagnosis. The spleen may become massively enlarged — sometimes extending into the pelvis — due to extramedullary hematopoiesis and leukemic cell infiltration.
- Abdominal fullness and early satiety: Massive splenomegaly compresses the stomach, reducing its capacity and producing a sensation of fullness with small meals. Patients may significantly reduce food intake, contributing to weight loss.
- Left upper quadrant pain and heaviness: Due to capsular distension of the enlarged spleen. Pain may be dull and persistent or sharp in nature.
- Splenic infarction: Acute, severe left upper quadrant pain radiating to the left shoulder (Kehr's sign) occurring when areas of the massively enlarged spleen undergo ischemic infarction due to impaired vascular supply. This can mimic pleuritis or acute abdomen.
- Referred left shoulder pain: Due to diaphragmatic irritation from large spleens pressing upward.
- Hepatomegaly: Less pronounced than splenomegaly but detectable in approximately 30–40% of patients due to hepatic infiltration by leukemic cells.
C. Hematological Manifestations
- Easy bruising and bleeding tendencies: Despite an often-elevated platelet count (reactive thrombocytosis) in chronic phase, platelet function is frequently abnormal, resulting in mucosal bleeding, petechiae, ecchymoses, and prolonged bleeding from minor wounds.
- Thrombotic complications: Elevated platelet counts and abnormal platelet activation may paradoxically increase thrombotic risk, including deep vein thrombosis, pulmonary embolism, and arterial events.
- Pallor: Clinical sign of anemia visible in conjunctivae, nail beds, and palmar creases. Reflects insufficient red cell production due to marrow infiltration.
- Hyperuricemia and gout: Massive cell turnover leads to excessive purine catabolism and uric acid production, potentially precipitating gouty arthritis or nephrolithiasis.
2. Accelerated Phase Manifestations
The accelerated phase is characterized by progressive disease that no longer responds adequately to prior treatment, with increasing blast percentage and accumulating cytogenetic abnormalities.
- Worsening constitutional symptoms: Fever, night sweats, and weight loss become more prominent and harder to control.
- Progressive splenomegaly: Spleen continues to enlarge despite treatment, reflecting increasing extramedullary disease burden.
- Increasing bone pain and joint pain: Due to expanding myeloid mass in the bone marrow, periosteal invasion, and bone remodeling.
- Worsening anemia and thrombocytopenia: Decreasing red cell and platelet production leads to increased fatigue, bleeding risk, and susceptibility to infection.
- Development of cytopenias refractory to treatment: A critical feature distinguishing accelerated phase from adequately controlled chronic phase disease.
- Lymphadenopathy: May appear or worsen as disease progresses, reflecting extramedullary leukemic infiltration of lymph nodes.
3. Blast Phase (Blast Crisis) Manifestations
Blast phase CML is a medical emergency and is clinically indistinguishable from de novo acute leukemia (AML in ~70% of cases; ALL in ~30%). Symptoms reflect acute bone marrow failure and rapid proliferation of blast cells.
- Severe constitutional symptoms: High-grade fever, drenching night sweats, profound weight loss, and extreme fatigue dominate the clinical picture.
- Severe bone pain: Marked tenderness on sternal pressure and over long bones due to massive marrow infiltration by blasts. Children may refuse to walk.
- Leukostasis syndrome: Occurs when the white blood cell count exceeds 100,000/µL. Hyperviscosity of the blood impairs microvascular flow, causing respiratory distress (pulmonary leukostasis), altered consciousness, visual disturbances, priapism, and digital ischemia. This is a life-threatening emergency requiring urgent cytoreduction.
- Bleeding diathesis: Severe thrombocytopenia combined with coagulation factor consumption may cause life-threatening hemorrhage including intracranial hemorrhage.
- Tumor lysis syndrome: Rapid destruction of blast cells releases massive quantities of intracellular contents — potassium, phosphate, uric acid, and nucleic acids — causing acute hyperkalemia, hyperphosphatemia, hyperuricemia, and hypocalcemia, with risk of acute kidney injury and fatal arrhythmias.
- Extramedullary blast accumulations (chloromas/granulocytic sarcomas): Localized collections of blast cells may appear in skin, lymph nodes, central nervous system, testes, or other sites, producing organ-specific symptoms such as proptosis, cranial nerve palsies, or skin nodules.
- CNS involvement: Meningeal leukemia may produce headache, nausea, photophobia, cranial nerve palsies, and papilledema.
Risk Factors of Chronic Myeloid Leukemia (CML)
Unlike many other malignancies, CML has relatively few established risk factors. The hallmark event — the chromosomal translocation t(9;22)(q34;q11.2) producing the Philadelphia chromosome — is an acquired somatic mutation that occurs in a single hematopoietic stem cell during a person's lifetime. It is not inherited in a Mendelian fashion. However, certain environmental exposures and individual characteristics have been associated with an increased risk of developing CML.
1. Ionizing Radiation Exposure
- High-dose ionizing radiation: The strongest and best-documented environmental risk factor for CML. Exposure to significant doses of ionizing radiation can induce double-strand DNA breaks, leading to chromosomal rearrangements including the t(9;22) translocation. The incidence of CML was significantly elevated among survivors of the atomic bomb explosions in Hiroshima and Nagasaki in 1945, with a dose-dependent relationship clearly demonstrated in epidemiological studies.
- Therapeutic radiation: Patients who have previously received radiation therapy for other malignancies (e.g., cervical cancer, ankylosing spondylitis, Hodgkin lymphoma) have an increased risk of developing CML years later, reflecting radiation-induced genomic instability in hematopoietic stem cells.
- Occupational radiation exposure: Radiologists, nuclear medicine workers, radiographers, and individuals working in nuclear power plants or fuel reprocessing facilities may have a marginally elevated risk, though modern radiation protection standards have dramatically reduced occupational exposure levels.
- Medical diagnostic radiation: Low-level diagnostic radiation from X-rays and CT scans is generally not considered to carry a clinically meaningful risk for CML at currently recommended exposure levels.
2. Age and Demographic Factors
- Advancing age: The risk of CML increases progressively with age, with the incidence rising steeply after the age of 50 years. Older individuals have accumulated more opportunities for somatic mutations in hematopoietic stem cells, and age-related decline in DNA repair mechanisms may contribute to susceptibility. The median age at diagnosis in Western countries is approximately 55–65 years.
- Male sex: Men have a modestly higher incidence of CML compared to women, with a male-to-female ratio of approximately 1.3–1.5:1. The biological basis for this gender disparity is not fully elucidated but may involve hormonal influences on hematopoiesis and immune surveillance.
- Pediatric and young adult CML: While CML accounts for only 2–3% of all childhood leukemias, it does occur in younger age groups and typically presents with more aggressive features. The molecular pathology is identical to adult CML, and TKI therapy is equally effective.
3. Genetic and Molecular Factors
- Philadelphia chromosome positivity: The BCR-ABL1 translocation is the defining genetic event in CML. Different BCR breakpoints produce different fusion protein isoforms (p210, p190, p230), which may influence disease phenotype and clinical behavior.
- Additional chromosomal abnormalities (ACAs): Patients who acquire additional chromosomal abnormalities such as trisomy 8, isochromosome 17q, or a second Philadelphia chromosome at diagnosis or during treatment may have a more aggressive disease course and increased risk of phase transformation.
- Absence of strong hereditary predisposition: CML does not follow a classical hereditary pattern. Familial clustering is rare, and first-degree relatives of CML patients do not have a substantially elevated risk compared to the general population.
4. Chemical and Occupational Exposures
- Benzene exposure: Chronic exposure to benzene, a known bone marrow toxin, has been associated with various hematological malignancies, including acute myeloid leukemia and, to a lesser extent, CML. Benzene-containing solvents are found in petroleum products, certain paints, adhesives, and rubber industries. Occupational exposure in petroleum refinery workers, shoe manufacturing, and printing has been implicated.
- Other chemical exposures: Alkylating agents used in cancer chemotherapy (e.g., busulfan, cyclophosphamide) may occasionally precipitate secondary leukemias, including CML-like disorders, though this is rare in clinical practice.
5. Host-Related Factors
- Immunocompromised state: While CML is not classically associated with immunodeficiency, impaired immune surveillance may theoretically reduce the body's ability to eliminate early leukemic clones, allowing their expansion.
- Prior hematological disorder: Patients with a history of myeloproliferative neoplasms or bone marrow failure syndromes may have an underlying susceptibility to clonal evolution in hematopoietic progenitors, though secondary CML is rare.
Diagnosis of Chronic Myeloid Leukemia (CML)
The diagnosis of CML integrates clinical evaluation, hematological investigation, morphological assessment of blood and bone marrow, and definitive molecular/cytogenetic confirmation. Because CML is definitively characterized by the Philadelphia chromosome and BCR-ABL1 fusion gene, molecular and cytogenetic testing is not merely confirmatory but forms the cornerstone of initial diagnosis, treatment selection, response monitoring, and resistance detection. A high index of clinical suspicion in any patient presenting with unexplained leukocytosis and splenomegaly is essential for timely diagnosis.
1. Clinical Assessment and Physical Examination
A. History Taking
- Symptom duration and progression: Establishing when fatigue, abdominal discomfort, early satiety, and constitutional symptoms began helps define phase of disease and pace of progression.
- History of radiation exposure: Therapeutic radiation history, occupational exposures, and prior chemotherapy are documented as potential contributing factors.
- Medication history: Certain drugs may cause reactive leukocytosis that must be differentiated from CML.
- Family history: While CML is not hereditary, a family history of hematological malignancies is noted.
B. Physical Examination
- Splenomegaly: Detected by percussion and palpation of the left upper quadrant. Size is documented in centimeters below the left costal margin. Massive splenomegaly (extending >10 cm below the costal margin) is characteristic of CML. A splenic rub may be heard in the presence of splenic infarction.
- Hepatomegaly: Present in approximately 30–40% of patients; detected by liver palpation and percussion.
- Sternal tenderness: Elicited on pressure over the lower sternum in patients with significant marrow infiltration. A characteristic sign of bone marrow disease.
- Pallor and signs of anemia: Conjunctival pallor, palmar crease pallor, and tachycardia may indicate significant anemia.
- Bleeding signs: Ecchymoses, petechiae, or purpura reflecting abnormal platelet function or thrombocytopenia.
- Lymphadenopathy: Typically absent in chronic phase but may be present in accelerated or blast phase.
2. Laboratory Diagnosis
A. Complete Blood Count (CBC) and Peripheral Blood Smear
The CBC is usually the first investigation to suggest CML and is frequently abnormal even in asymptomatic patients.
- Leukocytosis: The hallmark laboratory finding. WBC counts typically range from 20,000 to over 300,000 cells/µL (reference range: 4,500–11,000/µL). The degree of leukocytosis does not reliably predict disease phase but correlates with tumor burden.
- Left shift with myeloid spectrum: The peripheral blood smear in CML characteristically demonstrates a complete spectrum of myeloid maturation — from myeloblasts (usually <5% in CP), promyelocytes, myelocytes, metamyelocytes, band neutrophils, and mature segmented neutrophils — producing a pattern called a leukemoid reaction that mimics normal bone marrow morphology.
- Basophilia and eosinophilia: Absolute basophilia (>1% basophils) is an important diagnostic clue particularly helpful in distinguishing CML from benign leukemoid reactions. Eosinophilia is also frequently present.
- Thrombocytosis or thrombocytopenia: Platelet counts may be elevated (reactive thrombocytosis) in early chronic phase or reduced in advanced disease, reflecting marrow dysfunction.
- Anemia: Normocytic, normochromic anemia is common due to replacement of erythroid progenitors.
B. Leukocyte Alkaline Phosphatase (LAP) Score
- Low LAP score: In CML, the LAP score (a cytochemical measure of alkaline phosphatase activity in mature neutrophils) is characteristically very low or absent. This contrasts sharply with benign leukemoid reactions (e.g., severe infections), in which the LAP score is markedly elevated. This test, though largely replaced by molecular diagnostics in modern practice, remains a useful rapid bedside discriminator.
C. Bone Marrow Examination
Bone marrow aspiration and trephine biopsy provide essential morphological, histological, and prognostic information.
- Bone marrow aspiration: Reveals markedly hypercellular marrow (often 90–100% cellularity), with dominance of granulopoiesis showing cells at all stages of maturation. Megakaryocytes may be increased and are characteristically small with hypolobated nuclei (dwarf megakaryocytes). Blasts <10% in chronic phase.
- Trephine biopsy: Shows hypercellularity with marked myeloid expansion, reduced fat spaces, and often prominent reticulin fibrosis on silver staining. Reticulin fibrosis at diagnosis has prognostic significance.
- Blast percentage assessment: Critical for phase determination — <10% blasts: chronic phase; 10–19%: accelerated phase; ≥20%: blast phase (WHO criteria).
D. Cytogenetic Analysis — Conventional Karyotyping
- Philadelphia chromosome detection: Conventional cytogenetic analysis (G-banding karyotype) of bone marrow cells confirms the t(9;22)(q34;q11.2) translocation in >95% of CML cases. The karyotype also identifies additional chromosomal abnormalities (ACAs) that carry prognostic implications. Results typically take 10–14 days.
- Variant translocations: Approximately 5% of CML cases involve complex or variant translocations that may include one or more additional chromosomes, but the BCR-ABL1 fusion is still present and requires confirmation by molecular testing.
E. Fluorescence In Situ Hybridization (FISH)
- BCR-ABL1 FISH: A molecular cytogenetic technique that uses fluorescent DNA probes complementary to the BCR and ABL1 gene sequences to detect the fusion signal in interphase cells. FISH is faster than conventional karyotyping, applicable to both bone marrow and peripheral blood, and detects the translocation even in atypical or complex cases. It can detect BCR-ABL1 positivity in approximately 1 in 200 cells, making it more sensitive than conventional karyotyping.
F. Quantitative Real-Time PCR (RT-qPCR) — The Gold Standard for Monitoring
- BCR-ABL1 transcript quantification: RT-qPCR amplifies and quantifies BCR-ABL1 mRNA from peripheral blood or bone marrow and is the most sensitive molecular test available. Results are reported as a percentage on the International Scale (IS), where Major Molecular Response (MMR, also called MR3) corresponds to BCR-ABL1(IS) ≤0.1%. This technique is the cornerstone of treatment response monitoring and can detect 1 CML cell in 100,000 normal cells (10-log sensitivity).
- Transcript type identification: RT-PCR identifies the specific BCR-ABL1 transcript variant (most commonly e13a2 or e14a2, encoding p210), which is important because not all transcript types are quantifiable on the same IS scale.
G. BCR-ABL1 Kinase Domain Mutation Analysis
- Mutation screening: When patients develop resistance to TKI therapy, Sanger sequencing or next-generation sequencing of the BCR-ABL1 kinase domain detects resistance mutations such as T315I (the "gatekeeper" mutation, resistant to all first- and second-generation TKIs), F317L, E255K/V, Y253H, and others. The specific mutation identified guides selection of subsequent TKI therapy.
3. Biochemical and Ancillary Investigations
- Serum uric acid: Often elevated due to high cell turnover and purine catabolism. Monitoring prevents tumor lysis syndrome and gout.
- Lactate dehydrogenase (LDH): Elevated in CML, reflecting high cell turnover. Markedly elevated levels may suggest phase acceleration or blast crisis.
- Liver and renal function tests: Baseline hepatic and renal parameters are essential before TKI initiation, as these drugs undergo hepatic metabolism and may cause organ toxicity.
- Vitamin B12 levels: Markedly elevated serum vitamin B12 is characteristic of CML, resulting from overproduction of transcobalamin I by granulocytes. This finding may predate diagnosis and is a useful corroborating sign.
- Serum electrolytes: Pseudohyperkalemia may occur in CML due to in vitro leakage of potassium from the massively elevated white cell count in blood collection tubes.
- Coagulation profile: PT, aPTT, and fibrinogen are evaluated, particularly in blast phase where disseminated intravascular coagulation (DIC) may develop.
4. Prognostic Scoring at Diagnosis
Validated prognostic scoring systems stratify patients into risk groups and guide treatment intensity:
| Score | Variables Included | Risk Groups |
|---|---|---|
| Sokal Score | Age, spleen size, platelet count, blast percentage | Low / Intermediate / High |
| Euro (Hasford) Score | Sokal variables + eosinophil and basophil counts | Low / Intermediate / High |
| EUTOS Score | Basophil percentage, spleen size | Low / High |
| ELTS Score | Age, spleen size, platelet count, blast percentage | Low / Intermediate / High |
Complications of Chronic Myeloid Leukemia (CML)
Complications of CML arise from the direct consequences of uncontrolled myeloid proliferation and organ infiltration, from the hematological derangements of advanced disease, and from the adverse effects of TKI therapy. The most feared complication is disease progression to blast crisis — a transformation to acute leukemia with a drastically worsened prognosis. In the TKI era, most chronic phase patients do not progress to blast crisis if treated adequately, but complications from TKI toxicity, treatment resistance, and cardiovascular events have become increasingly important.
A. Disease-Related Complications
1. Blast Phase (Blast Crisis) Transformation
The most catastrophic complication of CML is transformation from chronic or accelerated phase to blast phase, representing the acquisition of additional genetic mutations that drive differentiation arrest and acute leukemia biology. Approximately 70% of blast crises are myeloid (resembling AML) and 30% are lymphoid (resembling ALL). Blast crisis may arise abruptly without a clear accelerated phase. Once blast crisis is established, median survival without effective rescue therapy is only 3–6 months. Combination of TKIs with acute leukemia chemotherapy regimens followed by allogeneic HSCT offers the best chance of re-establishing remission, but long-term outcomes remain poor.
2. Leukostasis Syndrome
A hematological emergency occurring when extreme leukocytosis (typically WBC >100,000/µL) causes blood hyperviscosity and microvascular obstruction. Affected organs include the lungs (respiratory failure, pulmonary infiltrates), brain (confusion, stroke, coma), retinae (visual disturbances), and peripheral vasculature (digital ischemia, priapism). Leukostasis carries a 20–40% early mortality risk and requires immediate management with hydroxyurea for rapid cytoreduction, supportive care, and leukapheresis in selected cases. Paradoxically, red cell transfusions may worsen hyperviscosity in this setting and should be used cautiously.
3. Splenic Complications
- Splenic infarction: Severe, acute left upper quadrant pain due to ischemic necrosis of segments of the massively enlarged spleen. Managed conservatively with analgesics and anticoagulation in most cases; rarely requires splenectomy.
- Splenic rupture: A rare but life-threatening emergency presenting with sudden severe abdominal pain, signs of internal hemorrhage, and hemodynamic instability. Requires emergency surgery.
- Hypersplenism: Excessive sequestration and destruction of blood cells by the enlarged spleen contributes to worsening cytopenias.
4. Tumor Lysis Syndrome (TLS)
Occurs when rapid destruction of a large number of leukemic cells — spontaneously or following initiation of cytoreductive therapy — releases massive quantities of intracellular contents into the bloodstream. This produces the metabolic tetrad of hyperuricemia, hyperphosphatemia, hyperkalemia, and hypocalcemia. Clinical consequences include acute kidney injury, cardiac arrhythmias (due to hyperkalemia), tetany and seizures (due to hypocalcemia), and potentially fatal metabolic acidosis. Prevention with adequate hydration, urinary alkalinization, and allopurinol or rasburicase prior to treatment initiation is mandatory in high-risk patients.
5. Thrombohemorrhagic Complications
- Arterial and venous thrombosis: Thrombocytosis, increased platelet activation, and systemic inflammation contribute to thrombotic events including stroke, myocardial infarction, deep vein thrombosis, and portal vein thrombosis, particularly in older patients with cardiovascular risk factors.
- Hemorrhage: Thrombocytopenia in advanced disease, combined with platelet dysfunction and coagulopathy (especially in blast phase), can cause life-threatening bleeding including intracranial hemorrhage, gastrointestinal bleeding, and pulmonary hemorrhage.
- Disseminated Intravascular Coagulation (DIC): May complicate blast phase CML, particularly lymphoid blast crisis, characterized by simultaneous activation of coagulation and fibrinolysis leading to both microvascular thrombosis and systemic bleeding.
6. Infections
Despite the quantitative excess of white blood cells in CML, the leukemic granulocytes are functionally defective and have impaired chemotaxis, phagocytosis, and killing capacity. Patients with advanced disease and profound neutropenia (paradoxically, in blast crisis where normal marrow function is suppressed) are particularly vulnerable to bacterial, fungal, and viral infections. Opportunistic infections become more prevalent with intensive chemotherapy regimens used in blast phase.
B. Treatment-Related Complications (TKI Adverse Effects)
Although TKIs have revolutionized CML management, they are not without adverse effects. Each TKI has a distinct toxicity profile, and long-term monitoring for specific complications is essential.
| TKI | Key Adverse Effects | Monitoring Required |
|---|---|---|
| Imatinib | Edema, nausea, muscle cramps, rash, fatigue, periorbital edema, hepatotoxicity | LFTs, CBC, weight |
| Dasatinib | Pleural effusion, pulmonary arterial hypertension, bleeding (platelet dysfunction), QT prolongation | Echo, chest X-ray, CBC, ECG |
| Nilotinib | Cardiovascular events (PAOD, MI, stroke), QT prolongation, hyperglycemia, pancreatitis, hepatotoxicity | ABI, lipids, glucose, ECG, LFTs |
| Bosutinib | Diarrhea, nausea, hepatotoxicity, rash, fluid retention | LFTs, CBC, GI symptoms |
| Ponatinib | Arterial occlusive events (high risk), pancreatitis, hepatotoxicity, hypertension, heart failure | Cardiovascular risk, LFTs, lipase |
TKI Resistance : Primary resistance (failure to achieve response milestones) or secondary resistance (loss of previously achieved response) may develop due to BCR-ABL1 kinase domain mutations (most importantly the T315I "gatekeeper" mutation), BCR-ABL1 gene amplification, clonal evolution with additional cytogenetic abnormalities, or BCR-ABL1-independent mechanisms. Resistance necessitates switching to an alternative generation TKI or, in T315I-positive patients, to ponatinib or asciminib (a STAMP inhibitor targeting the ABL1 myristoyl pocket), and potentially allogeneic HSCT evaluation.
Treatment of Chronic Myeloid Leukemia (CML)
The treatment of CML has undergone a revolutionary transformation since 2001 with the introduction of tyrosine kinase inhibitors (TKIs) targeting BCR-ABL1. Prior to TKI availability, treatment options included interferon-alpha, hydroxyurea, and allogeneic hematopoietic stem cell transplantation (allo-HSCT) — none of which reliably produced durable molecular remissions. The modern treatment algorithm is guided by molecular response milestones, with the goal of achieving and maintaining deep molecular responses (DMR), and ultimately achieving Treatment-Free Remission (TFR). The selection of specific TKIs, monitoring strategies, and escalation or de-escalation decisions require individualized assessment by hematology specialists.
A. Tyrosine Kinase Inhibitors (TKIs) — The Cornerstone of Therapy
1. First-Generation TKI: Imatinib Mesylate (Gleevec/Glivec)
Imatinib was the first clinically approved TKI and remains a cornerstone of CML therapy. It competitively inhibits the BCR-ABL1 tyrosine kinase by binding to the ATP-binding pocket of the kinase domain in its inactive conformation, preventing phosphorylation of downstream signaling substrates and thereby blocking proliferative and anti-apoptotic signals in CML cells.
- Standard dose: 400 mg orally once daily with food (to reduce gastrointestinal side effects).
- Efficacy: The landmark IRIS trial demonstrated that imatinib achieved complete cytogenetic response (CCyR) in ~87% of newly diagnosed chronic phase patients at 5 years, with an overall survival of ~89%.
- Key adverse effects: Edema (periorbital, pedal), nausea, diarrhea, muscle cramps, rash, fatigue, and hepatotoxicity. Most adverse effects are manageable with dose adjustment or supportive measures.
- Limitations: Resistance may develop due to BCR-ABL1 kinase domain mutations, and molecular response rates are lower and slower compared to second-generation TKIs.
2. Second-Generation TKIs: Dasatinib, Nilotinib, Bosutinib
Second-generation TKIs were developed to overcome imatinib resistance and induce faster, deeper molecular responses. They are 25–300 times more potent than imatinib against BCR-ABL1 and effective against most imatinib-resistant BCR-ABL1 mutations (except T315I).
- Dasatinib (Sprycel): 100 mg once daily. Binds BCR-ABL1 in both active and inactive conformations; also inhibits Src kinases. Key toxicity: pleural effusion, pulmonary arterial hypertension, bleeding risk.
- Nilotinib (Tasigna): 300 mg twice daily (front-line) or 400 mg twice daily (resistant/intolerant). Highly selective for BCR-ABL1 with minimal kinase off-target activity. Key toxicity: cardiovascular events (peripheral arterial occlusive disease, myocardial infarction, stroke), QT prolongation, hyperglycemia.
- Bosutinib (Bosulif): 400 mg (front-line) or 500 mg (resistant) once daily. Dual Src/ABL inhibitor. Key toxicity: diarrhea, nausea, elevated liver enzymes.
- Comparative efficacy: Second-generation TKIs achieve deeper molecular responses faster than imatinib and have higher rates of MR4 and MR4.5, facilitating earlier eligibility for TFR attempts.
3. Third-Generation TKI: Ponatinib (Iclusig)
- Indication: The only approved TKI active against the T315I "gatekeeper" mutation, which confers pan-resistance to all first- and second-generation TKIs. Also used in patients who have failed ≥2 prior TKIs.
- Dose: 45 mg once daily (can be reduced to 15 or 30 mg in responding patients to minimize cardiovascular toxicity).
- Key toxicity: Highest cardiovascular risk of all TKIs — arterial occlusive events (peripheral arterial disease, myocardial infarction, stroke, mesenteric ischemia) occur in up to 25% of patients. Requires careful cardiovascular risk stratification before use.
4. Asciminib (Scemblix) — STAMP Inhibitor
- Novel mechanism: Asciminib is the first approved TKI that targets the ABL1 myristoyl pocket (STAMP: Specifically Targeting the ABL Myristoyl Pocket) rather than the ATP-binding site, providing a distinct mechanism that overcomes resistance to ATP-competitive TKIs including the T315I mutation (at high doses).
- Indication: Approved for CML in chronic phase who have been previously treated with ≥2 TKIs; also active in T315I-positive patients at 200 mg twice daily dosing.
- Advantages: Favorable tolerability profile with fewer off-target kinase effects compared to ponatinib.
B. Treatment Response Milestones and Monitoring
Treatment response in CML is assessed according to predefined milestones using CBC, cytogenetics, and molecular testing. Failure to achieve response milestones at specified time points prompts treatment modification.
| Time Point | Optimal Response | Warning | Treatment Failure |
|---|---|---|---|
| 3 months | BCR-ABL1(IS) ≤10%, Ph+ <35% | BCR-ABL1(IS) >10% | No CHR, Ph+ >95% |
| 6 months | BCR-ABL1(IS) ≤1%, Ph+ 0% | BCR-ABL1(IS) 1–10% | BCR-ABL1(IS) >10%, Ph+ >35% |
| 12 months | BCR-ABL1(IS) ≤0.1% (MMR) | BCR-ABL1(IS) 0.1–1% | BCR-ABL1(IS) >1%, Ph+ >0% |
| Any time | MR4 (≤0.01%) / MR4.5 (≤0.0032%) | Loss of MMR | Loss of CHR, CCyR, or MMR |
CHR = Complete Hematologic Response; CCyR = Complete Cytogenetic Response; MMR = Major Molecular Response; MR4/MR4.5 = deep molecular responses
C. Treatment-Free Remission (TFR)
One of the most significant advances in modern CML management is the concept of Treatment-Free Remission — discontinuing TKI therapy in patients who have achieved sustained deep molecular response (MR4 or deeper for ≥2–3 years). Approximately 40–60% of eligible patients who discontinue TKI therapy maintain MMR or deeper responses without relapse. Patients who lose molecular response typically regain it rapidly upon TKI restart. TFR attempts are made under strict molecular monitoring protocols, and close follow-up is mandatory. TFR is considered the modern equivalent of a functional cure for CML.
D. Management of Special Situations
1. Accelerated Phase CML
- Switch to or dose-escalate to a second- or third-generation TKI if not already receiving one. Evaluate for allo-HSCT in eligible patients. Aim to return to chronic phase before proceeding to transplantation.
2. Blast Phase CML
- Initiate TKI therapy combined with acute leukemia induction chemotherapy (AML or ALL regimens depending on lineage). Consolidate with allo-HSCT in eligible patients achieving second chronic phase. Prognosis remains poor but responses are possible.
3. Allogeneic Hematopoietic Stem Cell Transplantation (Allo-HSCT)
- Indications in the TKI era: Allo-HSCT is no longer the first-line therapy for chronic phase CML but remains the only potentially curative intervention for TKI-resistant disease, blast phase CML unresponsive to TKIs, and patients with T315I mutation not amenable to available TKIs.
- Procedure: Involves myeloablative or reduced-intensity conditioning followed by infusion of matched sibling or unrelated donor hematopoietic stem cells. The graft-versus-leukemia (GvL) effect plays an important anti-leukemic role.
- Complications: Graft-versus-host disease (GvHD — acute and chronic), regimen-related toxicity, infections, secondary malignancies, and transplant-related mortality limit its application in older or comorbid patients.
4. CML in Pregnancy
- TKIs are teratogenic and must be discontinued before conception. Women with CML who wish to conceive require pre-conception counseling, TKI discontinuation (feasible only in deep molecular remission), and close molecular monitoring throughout pregnancy. Interferon-alpha may be used if cytoreductive therapy is needed during pregnancy. Hydroxyurea is avoided in the first trimester.
E. Supportive Therapy
- Allopurinol and hydration: Initiated before starting cytoreductive therapy to prevent tumor lysis syndrome and hyperuricemia in patients with high white cell counts.
- Hydroxyurea: A cytoreductive agent used for rapid initial reduction of extremely high white cell counts (leukocytosis control) while awaiting TKI therapy or in the perioperative period. Not disease-modifying and does not eradicate the CML clone.
- Leukapheresis: Used in cases of symptomatic leukostasis for rapid mechanical removal of leukemic cells from the blood. A temporary bridge measure that does not reduce the leukemic burden permanently.
- Growth factors: G-CSF may be considered in selected patients with severe TKI-induced neutropenia to support dose maintenance.
- Transfusion support: Red cell transfusions for symptomatic anemia; platelet transfusions for bleeding in thrombocytopenic patients with advanced disease.
Prevention and Control of Chronic Myeloid Leukemia (CML)
Unlike many infectious or lifestyle-related diseases, CML cannot currently be prevented in the absolute sense, because it arises from an acquired somatic chromosomal translocation — the BCR-ABL1 fusion — that occurs spontaneously and unpredictably in a single hematopoietic stem cell during a person's lifetime. There is no vaccine, no dietary supplement, and no pharmacological agent proven to prevent the development of CML in asymptomatic individuals. However, certain strategies can minimize exposure to known risk factors, facilitate early detection in high-risk populations, optimize treatment outcomes once the disease is diagnosed, and prevent disease progression and complications.
A. Primary Prevention: Minimizing Exposure to Known Risk Factors
1. Limiting Ionizing Radiation Exposure
Ionizing radiation is the only well-established environmental risk factor for CML. Minimizing unnecessary exposure to ionizing radiation is the single most important preventive strategy at the population level.
- Occupational radiation protection: Strict adherence to radiation safety principles — time, distance, and shielding — is mandatory for healthcare workers (radiologists, radiographers, nuclear medicine technicians), nuclear power plant workers, and others with occupational exposure. Regulatory bodies such as the International Commission on Radiological Protection (ICRP) and national radiation protection agencies set permissible annual dose limits that must be observed.
- Use of personal dosimeters: All radiation workers should wear personal dosimetry devices (film badges, thermoluminescent dosimeters, or electronic personal dosimeters) to monitor cumulative exposure and ensure limits are not exceeded.
- Judicious use of medical radiation: Clinicians should adhere to the ALARA (As Low As Reasonably Achievable) principle when ordering radiological investigations. CT scans and fluoroscopic procedures carry higher doses and should be ordered only when clinically indicated, with lower-dose alternatives (ultrasound, MRI) preferred when appropriate.
- Radiation therapy optimization: Radiation oncologists should use modern conformal and image-guided radiation therapy techniques to minimize dose to the bone marrow when treating malignancies, particularly in younger patients.
2. Minimizing Chemical Carcinogen Exposure
- Benzene exposure reduction: Workers in industries involving petroleum refining, rubber manufacturing, chemical synthesis, shoemaking, and printing should use appropriate personal protective equipment, ensure adequate workplace ventilation, and adhere to occupational exposure limit regulations. Benzene-containing products should be substituted with safer alternatives wherever possible.
- Smoking cessation: Although not directly linked to CML pathogenesis, tobacco smoke contains benzene and other myelotoxic carcinogens that may increase leukemia risk broadly. Smoking cessation programs are recommended for all patients with hematological malignancies and their at-risk family members.
B. Secondary Prevention: Early Detection and Timely Diagnosis
1. Recognition of Incidental Leukocytosis
Since approximately 40–50% of CML patients are asymptomatic at diagnosis and the disease is detected on routine CBC, primary care physicians and general practitioners must maintain vigilance for unexplained persistent leukocytosis in any adult patient. Any WBC count >20,000/µL in the absence of obvious infection, inflammation, or corticosteroid use warrants further investigation, including a peripheral blood smear, differential count, and referral to a hematologist for molecular testing.
2. Education of Healthcare Workers
Training primary care physicians, internists, and emergency physicians to recognize the clinical signs of CML — splenomegaly, unexplained fatigue, early satiety, and leukocytosis — facilitates early referral to hematology services, earlier initiation of TKI therapy, and prevention of phase progression that occurs in untreated or under-treated disease.
3. Monitoring of High-Risk Populations
- Individuals with a known history of significant radiation exposure (e.g., cancer survivors treated with radiotherapy, nuclear accident survivors) may benefit from periodic CBC monitoring to facilitate early detection of hematological abnormalities.
- Workers with chronic occupational benzene exposure should undergo regular hematological surveillance as part of occupational health programs.
C. Tertiary Prevention: Preventing Disease Progression and Complications
1. Adherence to TKI Therapy
Non-adherence to TKI therapy is one of the most important modifiable causes of treatment failure, suboptimal molecular response, and disease progression in CML patients. Studies have demonstrated that even minor reductions in TKI adherence (below 90%) are associated with significantly lower rates of molecular response. Healthcare teams should assess adherence at every visit, provide patient education about the importance of consistent medication use, and address barriers to adherence such as side effects, financial difficulties, and pill burden.
2. Regular Molecular Monitoring
BCR-ABL1 RT-PCR monitoring every 3 months allows detection of rising transcript levels that signal early loss of response, TKI resistance, or emerging kinase domain mutations — all before overt clinical or cytogenetic relapse occurs. Early intervention at the molecular level, rather than waiting for symptomatic relapse, significantly improves outcomes and reduces the risk of blast phase transformation.
3. Management of TKI Adverse Effects
Proactive identification and management of TKI side effects (edema with imatinib, pleural effusion with dasatinib, cardiovascular risk with nilotinib and ponatinib) prevents unnecessary dose reductions or discontinuations that could compromise molecular response. Cardiovascular risk factor management (hypertension, dyslipidemia, diabetes) is especially critical for patients receiving nilotinib or ponatinib.
4. Prevention of Infection
- Patients with CML — particularly those on intensive therapy or with blast phase disease — should receive appropriate vaccinations (influenza, pneumococcal, COVID-19) in accordance with immunocompromised host guidelines. Live attenuated vaccines are generally contraindicated during active treatment.
- Prophylactic antifungal and antibacterial therapy may be required in patients undergoing allo-HSCT or intensive chemotherapy for blast crisis.
5. Fertility Preservation
CML affects patients of reproductive age, and TKI therapy is associated with teratogenicity. Pre-treatment fertility counseling, sperm banking for male patients, and embryo or oocyte cryopreservation for female patients should be offered before initiating therapy where clinically feasible, to preserve future reproductive options.
D. Public Health and Research Strategies
- Access to TKI therapy: The dramatic improvement in CML survival achieved with TKIs is contingent on equitable global access to these medications. Generic imatinib has become widely available and has improved access in lower-income countries. Public health advocacy for affordable TKI access is an important component of population-level disease control.
- Patient registries and research databases: National and international CML patient registries (e.g., EUTOS registry in Europe) provide epidemiological data, facilitate clinical trial design, and monitor real-world treatment outcomes, driving continuous improvements in disease management.
- Research into CML stem cell eradication: Quiescent CML stem cells are relatively insensitive to TKI therapy and are believed to be the reservoir responsible for disease persistence and potential relapse after TKI discontinuation. Research into combination strategies targeting BCR-ABL1 and CML stem cell survival pathways (Hedgehog, Wnt, autophagy inhibition) aims to achieve complete eradication of the leukemic clone.
Common FAQs on Chronic Myeloid Leukemia (CML)
Bibliography on Chronic Myeloid Leukemia (CML)
- Williams Hematology – Editors: K. Kaushansky, M. A. Lichtman, J. T. Prchal, M. M. Levi, O. W. Press, L. J. Burns, and M. Caligiuri, Publisher: McGraw-Hill Education.
- Harrison's Principles of Internal Medicine – Editors: J. L. Jameson, A. S. Fauci, D. L. Kasper, S. L. Hauser, and J. Loscalzo, Publisher: McGraw-Hill Education.
- Hoffbrand's Essential Haematology – Authors: A. V. Hoffbrand and D. P. Steensma, Publisher: Wiley-Blackwell.
- Clinical Hematology – Editors: N. S. Young, S. L. Gerson, and K. A. High, Publisher: Mosby Elsevier.
- Wintrobe's Clinical Hematology – Editors: J. P. Greer, D. A. Arber, B. Glader, A. D. List, R. T. Means, and F. Paraskevas, Publisher: Lippincott Williams & Wilkins.
- Cancer: Principles and Practice of Oncology – Editors: V. T. DeVita Jr., T. S. Lawrence, and S. A. Rosenberg, Publisher: Lippincott Williams & Wilkins.
- De Vita, Hellman, and Rosenberg's Cancer: Principles and Practice of Oncology – Editors: V. T. DeVita and T. S. Lawrence, Publisher: Wolters Kluwer.
- Dasatinib versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia (DASISION Trial) – Authors: H. M. Kantarjian et al., Journal: New England Journal of Medicine.
- Nilotinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia (ENESTnd) – Authors: R. A. Larson et al., Journal: New England Journal of Medicine.
- Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia (IRIS Trial) – Authors: B. J. Druker et al., Journal: New England Journal of Medicine.
- Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia – Authors: B. J. Druker et al., Journal: New England Journal of Medicine.
- Treatment-Free Remission in Patients with Chronic Myeloid Leukemia in Deep Molecular Response (EURO-SKI) – Authors: S. Mahon et al., Journal: Lancet Oncology.
- European LeukemiaNet Recommendations for the Management of Chronic Myeloid Leukemia – Authors: A. Hochhaus et al., Journal: Leukemia.
- Ponatinib in Patients with Philadelphia Chromosome-Positive Leukemias Resistant to Dasatinib or Nilotinib – Authors: H. M. Kantarjian et al., Journal: New England Journal of Medicine.
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues – Editors: S. H. Swerdlow, E. Campo, N. L. Harris, et al., Publisher: International Agency for Research on Cancer (IARC), Lyon.