

Acute Myeloid Leukemia (AML) – Sign and Symptoms, Risk Factors, Diagnosis, Complications, Treatment and Prevention
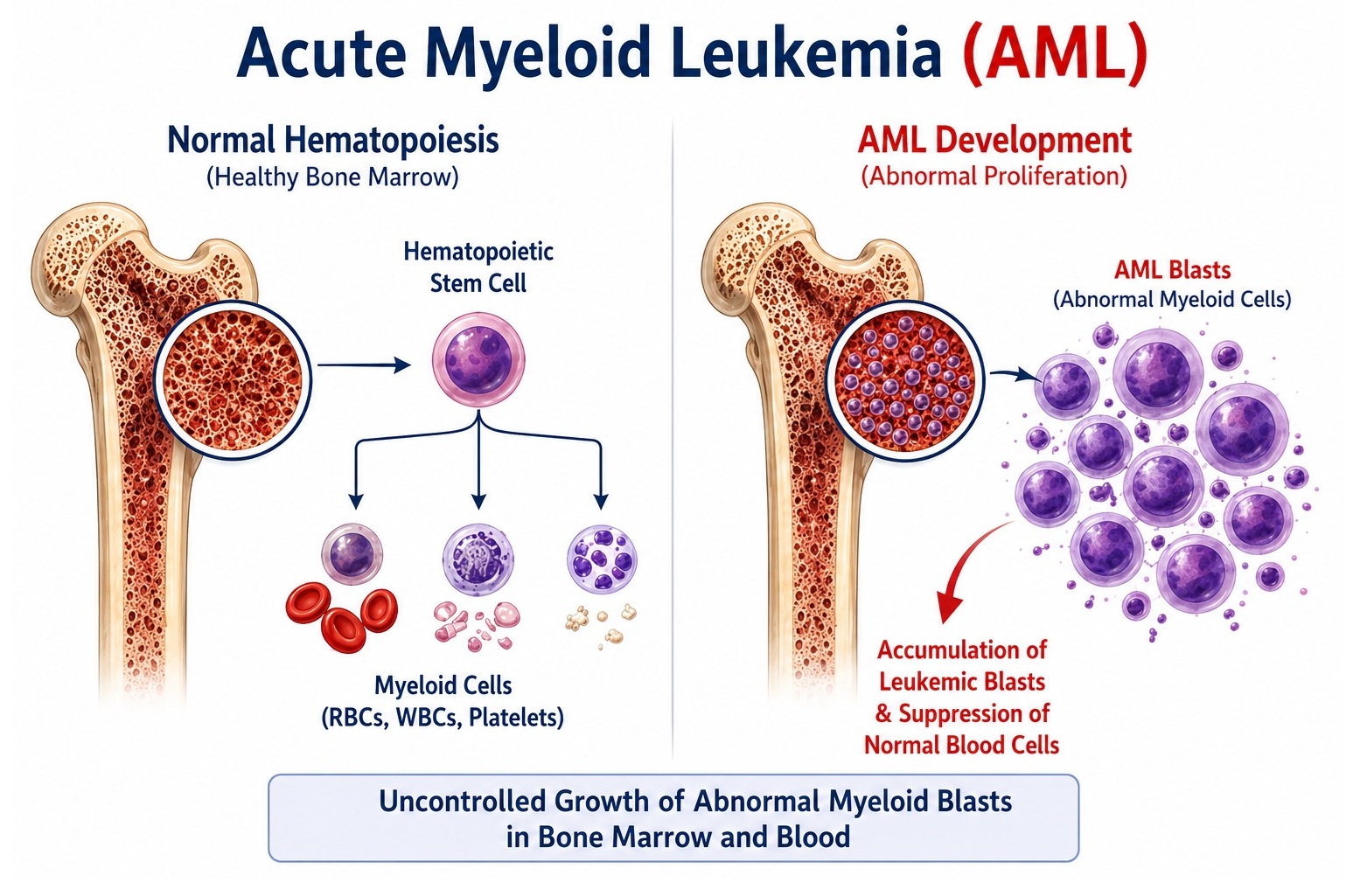
Pathologically, AML is defined by the presence of ≥20% blasts in the bone marrow or peripheral blood (WHO 2016/2022 criteria), or by specific recurrent cytogenetic and molecular abnormalities regardless of blast count. The disease encompasses a clinically and biologically heterogeneous group of subtypes, broadly classified into AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes, therapy-related AML, and AML not otherwise specified. Blasts accumulate in the marrow, crowding out erythroid, myeloid, and megakaryocytic precursors, resulting in anemia, neutropenia, and thrombocytopenia — the triad that underlies the primary clinical manifestations of fatigue, susceptibility to infection, and bleeding.
Initial evaluation relies on complete blood count, peripheral blood smear, bone marrow aspiration and biopsy, cytogenetics (karyotype), and comprehensive molecular profiling, collectively informing prognosis and guiding therapeutic decision-making. Standard induction therapy involves cytarabine-based regimens combined with anthracyclines. Targeted agents — including FLT3 inhibitors (midostaurin, gilteritinib), IDH1/2 inhibitors (ivosidenib, enasidenib), and BCL-2 inhibitor venetoclax — have significantly expanded treatment options. Allogeneic hematopoietic stem cell transplantation remains the primary curative consolidation strategy for eligible patients with intermediate- and high-risk disease. Despite advances, overall five-year survival remains approximately 30%, underscoring the continued need for novel therapeutic strategies and risk-adapted treatment protocols.
Classification of Acute Myeloid Leukemia
AML is classified according to the WHO 2022 Classification of Haematopoietic Tumours and the parallel International Consensus Classification (ICC), both of which incorporate morphology, cytogenetics, and molecular genetics:
| WHO 2022 Category | Key Defining Feature |
|---|---|
| AML with defining genetic abnormalities | t(8;21), inv(16), t(15;17) [APL], NPM1, CEBPA, FLT3-ITD, IDH1/2, KMT2A rearrangement, etc. |
| AML, myelodysplasia-related | Prior MDS/MPN, MDS-related cytogenetics or somatic mutations (SRSF2, SF3B1, ASXL1, etc.) |
| Therapy-related myeloid neoplasm | Prior cytotoxic chemotherapy or radiation therapy |
| AML, not otherwise specified (NOS) | Does not meet criteria for above; classified by morphological differentiation (FAB-inspired) |
| Acute promyelocytic leukemia (APL) | t(15;17); PML-RARA fusion — treated distinctly with ATRA + arsenic |
| Blastic plasmacytoid dendritic cell neoplasm | CD4+/CD56+ blasts; BDCA-2 positivity |
The older FAB (French-American-British) classification (M0–M7) remains useful in resource-limited settings and is referenced in pathology practice. The WHO system supersedes it for clinical decision-making by incorporating molecular and cytogenetic prognostic data.
Signs and Symptoms of Acute Myeloid Leukemia
AML typically presents acutely or subacutely over days to weeks. Symptoms arise from two principal pathological processes: (1) bone marrow failure due to replacement of normal hematopoiesis by leukemic blasts, and (2) leukemic infiltration of tissues. Clinical features may initially resemble other hematological conditions, necessitating a high index of suspicion.
Key clinical triad of AML:
- Anemia (fatigue, pallor, dyspnea) — from erythroid suppression
- Thrombocytopenia (bleeding, bruising) — from megakaryocytic suppression
- Neutropenia (recurrent infections, fever) — from mature granulocyte depletion

1. Constitutional and Systemic Symptoms
Constitutional symptoms often precede the diagnosis and reflect systemic effects of rapidly proliferating leukemic cells and associated cytokine dysregulation:
- Fatigue and generalized weakness: The most common presenting symptom, resulting from normochromic normocytic anemia due to suppressed red cell production in the marrow. Patients report diminished stamina, decreased exercise tolerance, and persistent tiredness unrelieved by rest.
- Pallor: A clinical manifestation of anemia, most evident on examination of the conjunctivae, nail beds, and palmar creases.
- Fever: May occur due to neutropenic sepsis, leukemia-associated pyrexia from cytokine release (IL-1, IL-6, TNF-α), or both. Fever in the context of newly diagnosed AML must always raise suspicion of an underlying infection requiring immediate evaluation.
- Night sweats and weight loss: Systemic tumor burden promotes a hypermetabolic state with increased energy expenditure and catabolic cytokine activity, resulting in anorexia and progressive weight loss.
- Bone and joint pain: Periosteal infiltration by leukemic blasts or direct marrow expansion produces diffuse bony tenderness, most pronounced in the sternum, ribs, and long bones. This is particularly notable in children with AML.
2. Hemorrhagic Manifestations
Bleeding complications arise from thrombocytopenia, dysfunctional platelets, and — critically in Acute Promyelocytic Leukemia (APL) — disseminated intravascular coagulation (DIC):
- Petechiae and purpura: Small, pinpoint, non-blanching hemorrhagic spots appearing most commonly on the lower limbs, representing capillary hemorrhage from platelet counts below 20–30 × 10⁹/L.
- Easy bruising (ecchymosis): Subcutaneous bleeding following minimal trauma, often at pressure points and injection sites.
- Mucosal bleeding: Gingival bleeding, epistaxis, and menorrhagia are among the early and frequently encountered bleeding manifestations prompting medical consultation.
- Prolonged bleeding from minor cuts: Impaired primary hemostasis due to low platelet count and qualitative platelet dysfunction.
- Serious hemorrhage: Intracranial hemorrhage, gastrointestinal bleeding, and pulmonary hemorrhage represent life-threatening complications, particularly in APL-associated DIC or with extreme thrombocytopenia.
3. Infectious Manifestations
Neutropenia from marrow failure renders patients profoundly immunocompromised and vulnerable to bacterial, fungal, and opportunistic infections:
- Febrile neutropenia: Defined as fever ≥38.3°C in a patient with absolute neutrophil count (ANC) <500/μL (or ANC <1000/μL expected to fall below 500/μL). Represents an oncologic emergency requiring empiric broad-spectrum antibiotic therapy.
- Recurrent bacterial infections: Gram-negative bacilli (Pseudomonas aeruginosa, Escherichia coli, Klebsiella) and gram-positive cocci (Staphylococci, Streptococci) frequently cause bacteremia and septic shock in neutropenic patients.
- Invasive fungal infections: Aspergillus and Candida species are the predominant pathogens in prolonged neutropenic states. Invasive pulmonary aspergillosis carries high mortality and requires early antifungal therapy.
- Oral mucositis and pharyngitis: Neutropenia impairs mucosal immunity, promoting development of oral ulceration, candidiasis, and herpes simplex virus reactivation in the oropharynx.
- Perianal infections: Due to mucosal breakdown and altered gut flora, perirectal cellulitis and abscesses may develop, particularly with prolonged neutropenia.
4. Leukostasis and Hyperleukocytosis
When the peripheral blast count exceeds 100,000/μL, leukostasis — a medical emergency — may occur due to intravascular aggregation of blasts in microcirculation, impairing tissue oxygenation:
- Pulmonary leukostasis: Dyspnea, hypoxemia, diffuse bilateral infiltrates on chest imaging, and respiratory failure from blast-occluded pulmonary capillaries.
- Cerebral leukostasis: Altered mental status, headache, blurred vision, focal neurological deficits, papilledema, and intracranial hemorrhage due to blast plugging in cerebral microvasculature.
- Priapism: Painful sustained penile erection from blast-related vascular occlusion in penile sinusoids — a rare but recognized feature.
- Retinal hemorrhages and visual disturbance: Identified on fundoscopic examination in patients with extreme hyperleukocytosis.
5. Organomegaly and Extramedullary Involvement
- Hepatosplenomegaly: Leukemic infiltration of the liver and spleen produces organomegaly, occasionally causing abdominal discomfort, early satiety, and pain from splenic infarction or rupture.
- Lymphadenopathy: Less common in AML than ALL but may occur in subtypes with monocytic differentiation (AML-M4/M5), reflecting tissue tropism of monocytic blasts.
- Gingival hypertrophy: A distinctive feature of AML subtypes with monocytic differentiation (particularly AML-M4 and AML-M5), caused by leukemic infiltration of gingival tissue producing characteristic bluish-red gingival overgrowth.
- Skin infiltration (Leukemia cutis): Violaceous, non-tender nodular or plaque-like dermal infiltrates representing extramedullary AML deposits. More common in monocytic subtypes and AML with KMT2A rearrangements.
- Chloroma (Granulocytic sarcoma / Myeloid sarcoma): Tumoral mass composed of leukemic myeloblasts at extramedullary sites — orbit, spine, breast, soft tissue — which may precede overt AML by weeks to months.
- Central nervous system involvement: Uncommon at diagnosis (<5%), but may present with cranial nerve palsies, meningismus, or elevated CSF blast count. More frequent in monocytic subtypes and pediatric AML.
- Testicular infiltration: Rarely observed; presents as painless testicular swelling.
6. Metabolic and Laboratory-Evident Symptoms
- Tumor lysis syndrome (TLS): Spontaneous or chemotherapy-induced rapid cell lysis releases large amounts of uric acid, potassium, and phosphate, leading to hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia, and acute kidney injury. Patients may present with tetanic cramps, cardiac arrhythmias, oliguria, and seizures.
- Hyperuricemia: Reflects elevated nucleic acid turnover from rapid leukemic proliferation; may manifest as gout-like arthropathy or uric acid nephropathy.
- Hyperviscosity: In hyperleukocytosis, the increased blood viscosity from circulating blasts contributes to microvascular sludging and compromised organ perfusion.
- Coagulopathy (DIC/ATRA-associated): Particularly in APL; patients present with systemic bleeding, hypofibrinogenemia, elevated D-dimer, and prolonged PT/aPTT — a direct result of tissue factor and procoagulant granule release from promyelocytes.
Risk Factors of Acute Myeloid Leukemia
AML risk is determined by an interplay of genetic predispositions, environmental exposures, prior hematological conditions, and iatrogenic factors. The majority of AML cases arise de novo without an identifiable antecedent cause; however, the following risk factors are well-established: 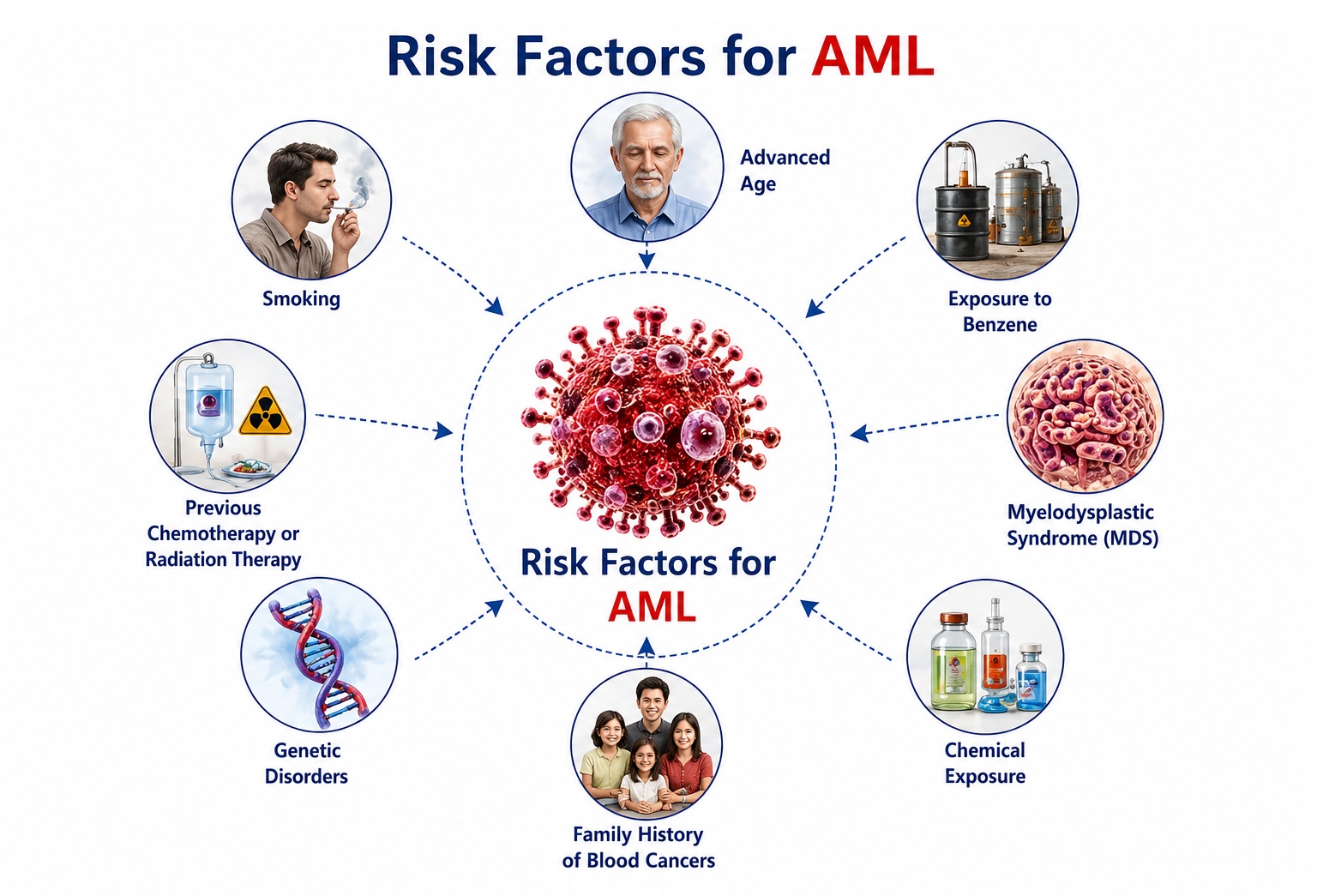
1. Age and Gender
- Advanced age: The most significant risk factor. AML incidence rises sharply with age; the median diagnosis age is approximately 68 years. Accumulation of somatic mutations and clonal hematopoiesis of indeterminate potential (CHIP) with aging underlies this association.
- Male sex: AML is slightly more common in males than females, with an estimated male-to-female ratio of 1.3:1. Hormonal, occupational, and genetic differences may contribute.
2. Genetic and Hereditary Predisposition
- Down syndrome (Trisomy 21): Associated with a markedly elevated risk of AML, particularly acute megakaryoblastic leukemia (AMKL), in children under 5 years. GATA1 somatic mutations are a hallmark of Down syndrome-associated AML.
- Fanconi anemia: An inherited DNA repair disorder caused by biallelic mutations in FANC genes, associated with extremely high risk of AML and other myeloid malignancies due to impaired genomic stability.
- Bloom syndrome: BLM gene mutations cause chromosomal instability, predisposing to multiple malignancies including AML.
- Diamond–Blackfan anemia, Schwachman–Diamond syndrome, and severe congenital neutropenia (Kostmann syndrome): These inherited bone marrow failure syndromes carry measurable risk of evolution to AML, reflecting clonal selection under conditions of chronic hematopoietic stress.
- Germline mutations: Hereditary mutations in RUNX1, CEBPA, GATA2, ETV6, DDX41, and TP53 have been recognized as predisposing to familial AML and myeloid malignancies. Genetic counseling is recommended for affected families.
- Clonal hematopoiesis of indeterminate potential (CHIP): Somatic mutations in DNMT3A, TET2, ASXL1 acquired in aging hematopoietic stem cells, without overt hematological malignancy, confer a significantly elevated AML risk.
3. Prior Hematological Disorders
- Myelodysplastic syndrome (MDS): A substantial proportion of AML cases — particularly therapy-related AML and AML with myelodysplasia-related changes — evolve from antecedent MDS. The risk is highest in high-grade MDS (RAEB-1, RAEB-2).
- Myeloproliferative neoplasms (MPNs): Polycythemia vera, essential thrombocythemia, and primary myelofibrosis may transform to AML (blast-phase MPN), a particularly aggressive disease with poor prognosis.
- Aplastic anemia: Treated aplastic anemia carries a risk of late-onset clonal evolution to MDS or AML.
4. Prior Cytotoxic Therapy (Therapy-Related AML)
- Alkylating agents: Cyclophosphamide, melphalan, busulfan, and other alkylators cause DNA crosslinking and strand breaks that can induce secondary chromosomal abnormalities (del(5q), del(7q), complex karyotype), typically manifesting as therapy-related MDS/AML 5–10 years after treatment.
- Topoisomerase II inhibitors: Etoposide, doxorubicin, and epirubicin cause DNA double-strand breaks that predispose to balanced translocations involving KMT2A (11q23) and other oncogenes, typically presenting as therapy-related AML within 1–3 years with balanced translocations.
- Radiation therapy: Prior therapeutic ionizing radiation, particularly to the pelvis or chest, contributes to DNA damage in hematopoietic progenitors and increases AML risk.
5. Environmental and Occupational Exposures
- Benzene: A well-established human leukemogen classified as Group 1 carcinogen by IARC. Chronic occupational exposure in the petroleum, rubber, chemical, and shoe manufacturing industries significantly increases AML risk by causing clastogenic DNA damage in hematopoietic stem cells.
- Tobacco smoking: Cigarette smoke contains benzene, formaldehyde, and other leukemogens. Smokers have approximately 1.4–2.0-fold increased risk of AML compared to non-smokers. The association is strongest for AML with chromosome 7 abnormalities.
- Pesticides and herbicides: Occupational or residential exposure to certain organochlorine pesticides and herbicides has been associated with elevated AML risk in epidemiological studies.
- Ionizing radiation: High-dose radiation exposure (atomic bomb survivors, nuclear accident victims) dramatically increases AML risk in a dose-dependent manner. Medical radiation from diagnostic studies confers minimal risk.
- Hair dye use: Long-term personal hair dye use, particularly permanent dark dyes containing aromatic amines, has been associated with a modest increase in leukemia risk in some studies.
6. Other Associations
- Obesity: Higher body mass index (BMI) has been identified as an independent risk factor for AML in prospective cohort studies, possibly through adipokine-mediated effects on hematopoietic progenitors and systemic inflammation.
- Viral infections: Human T-cell lymphotropic virus (HTLV-1) is implicated in adult T-cell leukemia/lymphoma; Epstein-Barr virus may rarely contribute to myeloid transformation in immunosuppressed individuals, though direct AML causality remains less established than for lymphoid malignancies.
- Electromagnetic fields: Extremely low-frequency electromagnetic field exposure has been evaluated as a risk factor with inconclusive evidence; associations reported in some childhood leukemia studies require further investigation.
Diagnosis of Acute Myeloid Leukemia
Diagnosis of AML requires integration of clinical features, morphological analysis, immunophenotyping, cytogenetic testing, and molecular profiling. All newly diagnosed AML requires comprehensive evaluation for risk stratification and treatment planning according to ELN 2022 (European LeukemiaNet) or NCCN guidelines. 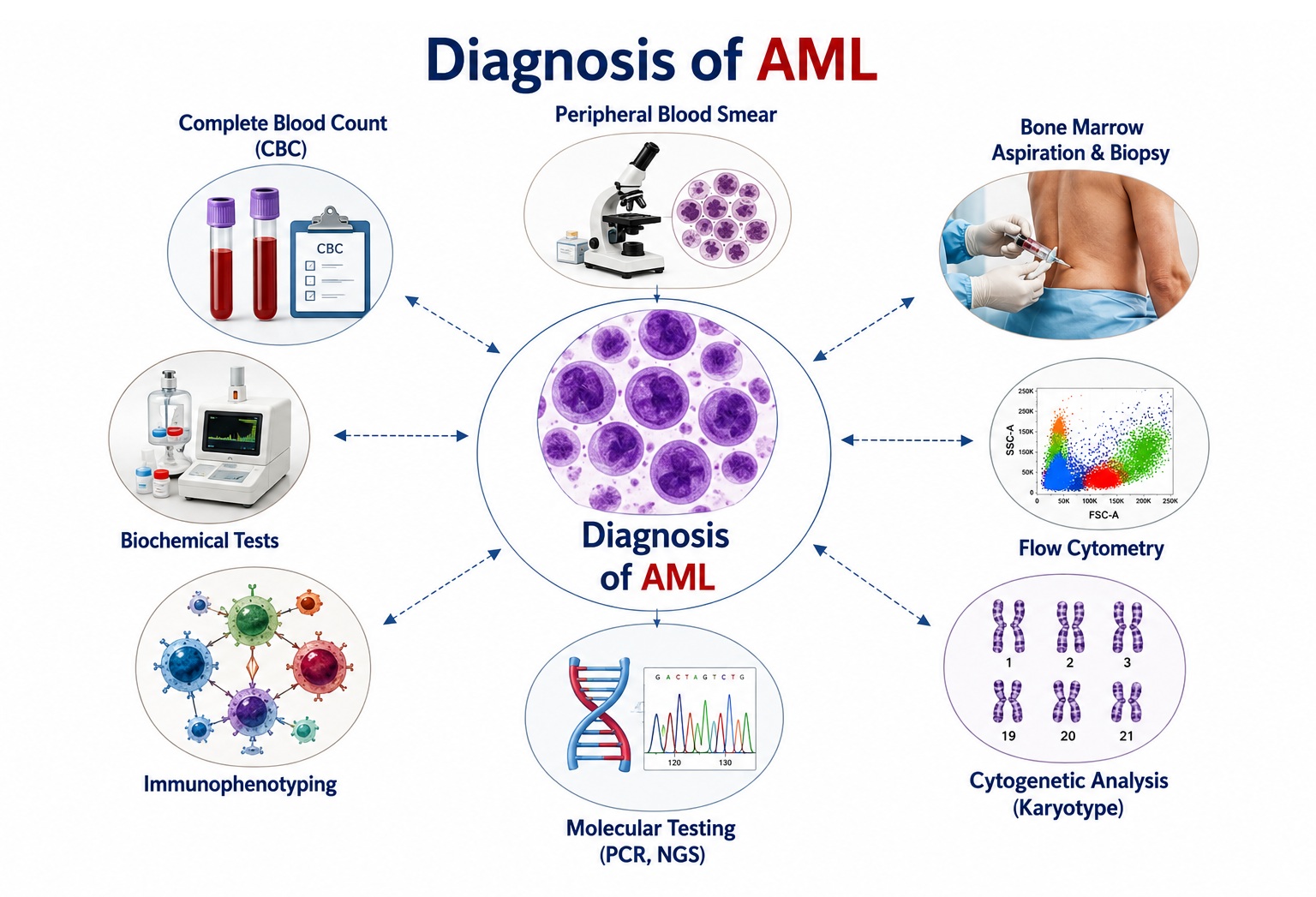
1. Clinical Examination
A. General Physical Examination
- General appearance: Pallor, cachexia, and fatigue reflect underlying anemia and hypermetabolic state. Dyspnea on exertion from anemia, and icterus if hemolytic components are present.
- Vital signs: Fever (febrile neutropenia), tachycardia, tachypnea indicating infection or anemia. Hypotension may suggest septic shock — a medical emergency requiring immediate intervention.
- Lymph nodes: Regional or generalized lymphadenopathy, more common in monocytic subtypes.
- Skin examination: Petechiae, purpura, ecchymoses reflecting thrombocytopenia. Leukemia cutis (dermal nodules/plaques). Pallor of skin and mucous membranes from anemia.
- Oral cavity: Gingival hypertrophy — pathognomonic of monocytic AML; oral ulceration, candidiasis reflecting neutropenic mucositis.
B. Abdominal Examination
- Hepatomegaly: Palpable liver edge below the costal margin due to leukemic infiltration.
- Splenomegaly: Palpable or percussable enlarged spleen, variable in extent; may cause left upper quadrant discomfort. Splenic infarction may cause acute pain.
C. Neurological Examination
- Assessment for cranial nerve palsies, meningismus, and papilledema in patients with headache or visual disturbance; CNS AML is uncommon but requires prompt evaluation.
- In hyperleukocytosis: altered sensorium, confusion, focal deficits from cerebral leukostasis.
2. Laboratory Investigations
A. Complete Blood Count (CBC) and Peripheral Blood Smear
- Anemia: Normochromic normocytic anemia; hemoglobin typically <10 g/dL at presentation.
- Thrombocytopenia: Platelet count usually <100 × 10⁹/L; severe (<20 × 10⁹/L) predisposes to spontaneous hemorrhage.
- WBC count: Variable — may be low (leukopenia), normal, or markedly elevated (hyperleukocytosis >100 × 10⁹/L). Circulating blasts may be seen on the differential count.
- Peripheral smear: Presence of myeloblasts with or without Auer rods (pathognomonic linear azurophilic cytoplasmic inclusions composed of fused primary granules, particularly seen in AML-M2 and APL). Auer rods confirm myeloid lineage.
B. Bone Marrow Aspiration and Biopsy
The cornerstone of AML diagnosis. Essential for morphological classification, immunophenotyping, cytogenetics, and molecular testing:
- Bone marrow aspirate: Demonstrates ≥20% myeloblasts (by WHO 2022) with morphological assessment of blast lineage and dysplastic changes across hematopoietic lineages.
- Trephine biopsy: Assesses marrow cellularity, blast distribution, reticulin fibrosis, and architecture.
- Cytochemistry: Myeloperoxidase (MPO) and Sudan Black B positivity confirm myeloid differentiation; non-specific esterase (NSE) positivity indicates monocytic differentiation.
C. Immunophenotyping (Flow Cytometry)
Multi-parameter flow cytometry defines the blast phenotype by identifying cell-surface and intracellular markers. Key markers include:
- Myeloid markers: CD13, CD33, CD117 (c-KIT), CD34, MPO (cytoplasmic)
- Monocytic markers: CD14, CD64, CD11b, NSE
- APL markers: CD33+, CD13+, HLA-DR negative, CD34 negative — a highly characteristic immunophenotype
- Megakaryoblastic markers: CD41, CD42, CD61 (for AMKL / AML-M7)
- Aberrant markers: Identification of anomalous antigen expression patterns (e.g., lymphoid antigens on myeloid blasts) is important for minimal residual disease (MRD) monitoring
3. Cytogenetic Analysis
A. Conventional Karyotyping (G-banding)
Analysis of metaphase chromosomes from bone marrow blasts identifies structural and numerical chromosomal abnormalities that are critical for risk stratification:
| Cytogenetic Abnormality | ELN 2022 Risk Group | Clinical Significance |
|---|---|---|
| t(8;21)(q22;q22); RUNX1-RUNX1T1 | Favorable | High CR rate; HDAC consolidation adequate |
| inv(16)(p13.1q22) or t(16;16); CBFB-MYH11 | Favorable | Good prognosis with cytarabine-based therapy |
| t(15;17)(q24;q21); PML-RARA (APL) | Favorable | ATRA + ATO curative in most patients |
| Normal karyotype | Intermediate (depends on molecular) | Prognosis defined by NPM1, FLT3, CEBPA status |
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | Intermediate | Monocytic differentiation common |
| Monosomy 5, del(5q), monosomy 7 | Adverse | Poor prognosis; allo-SCT in CR1 recommended |
| Complex karyotype (≥3 abnormalities) | Adverse | Very poor prognosis; often therapy-related |
| Monosomal karyotype | Adverse | Extremely poor outcome |
B. FISH (Fluorescence In Situ Hybridization)
Used to detect specific translocations and deletions not visible on conventional karyotyping; particularly useful for rapid confirmation of APL (PML-RARA) and KMT2A rearrangements.
4. Molecular Genetic Testing
Comprehensive molecular profiling is mandatory for all newly diagnosed AML to define prognosis, identify targetable mutations, and guide therapy selection:
- NPM1 mutations: Present in ~30% of AML; favorable prognosis in the absence of FLT3-ITD co-mutation; defines a distinct WHO entity.
- FLT3-ITD and FLT3-TKD: FLT3-ITD (~25%) confers adverse prognosis; FLT3 inhibitors (midostaurin, gilteritinib, quizartinib) are used in treatment. Allelic ratio and co-mutations modify prognosis.
- IDH1 and IDH2: Found in ~10% and ~15% of AML respectively; associated with leukemogenesis via 2-hydroxyglutarate accumulation; targetable with ivosidenib (IDH1) and enasidenib/olutasidenib (IDH2).
- CEBPA: Biallelic CEBPA mutations confer favorable prognosis; monoallelic mutations are less significant prognostically.
- RUNX1, ASXL1, TP53: Associated with adverse prognosis; TP53 mutations, particularly biallelic, carry the poorest outcomes and may respond to decitabine-based regimens.
- KIT mutations: In core-binding factor (CBF) AML with t(8;21) or inv(16), KIT D816 mutations may confer worse prognosis.
- DNMT3A, TET2, EZH2: Epigenetic regulators involved in MDS evolution to AML; prognostic and clinically relevant.
5. Minimal Residual Disease (MRD) Assessment
MRD monitoring is essential for evaluating treatment response and predicting relapse risk. Methods include:
- Multiparameter flow cytometry (MFC): Detection of aberrant leukemia-associated immunophenotype (LAIP) at sensitivity levels of 10⁻⁴–10⁻⁵.
- Quantitative PCR (qPCR): Highly sensitive MRD detection for specific molecular targets (NPM1, RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA) at sensitivities of 10⁻⁵–10⁻⁶.
- Next-generation sequencing (NGS)-based MRD: Increasingly used for patients without standard PCR markers; broader applicability across mutation types.
6. Additional Investigations
- Biochemistry panel: LFTs, RFTs, electrolytes, LDH (elevated in rapid cell turnover), uric acid, and coagulation studies (PT, aPTT, fibrinogen, D-dimer — essential to detect DIC, especially in APL).
- HLA typing: Initiated early for patients who may be candidates for allogeneic stem cell transplantation.
- Lumbar puncture (LP): For CSF cytology and immunophenotyping when CNS AML is suspected; not routinely performed at diagnosis unless neurological symptoms are present.
- CT scan / PET-CT: Evaluation of organomegaly, extramedullary AML (chloroma/myeloid sarcoma), lymphadenopathy, and infection screening pre-chemotherapy.
- Echocardiography: Mandatory before anthracycline-based induction to assess baseline cardiac function (LVEF); important in elderly patients.
Complications of Acute Myeloid Leukemia
Complications in AML arise from the disease itself, from the toxicity of chemotherapy and targeted therapies, and from the immunosuppressed state that accompanies treatment. Early recognition and management of complications are pivotal to improving survival and quality of life:
A. Disease-Related Complications
1. Sepsis and Infections
Febrile neutropenia and sepsis are the most life-threatening complications during AML treatment. Gram-negative bacteremia (Pseudomonas, Klebsiella, E. coli), gram-positive bacteremia (Staphylococci, Streptococci, Enterococci), and fungal infections (invasive aspergillosis, candidemia) are the most common causative organisms. Septic shock may develop rapidly, requiring immediate empiric antibiotic therapy, hemodynamic resuscitation, and ICU-level care.
2. Disseminated Intravascular Coagulation (DIC)
DIC is a hallmark of APL (AML-M3) and may occur in other AML subtypes during initial treatment or sepsis. Uncontrolled activation of both coagulation and fibrinolysis results in simultaneous thrombosis and hemorrhage. Laboratory hallmarks include prolonged PT/aPTT, low fibrinogen, elevated D-dimer, and thrombocytopenia. DIC in APL is an oncologic emergency mandating immediate ATRA initiation prior to or concurrent with chemotherapy, with close monitoring and aggressive transfusional support. 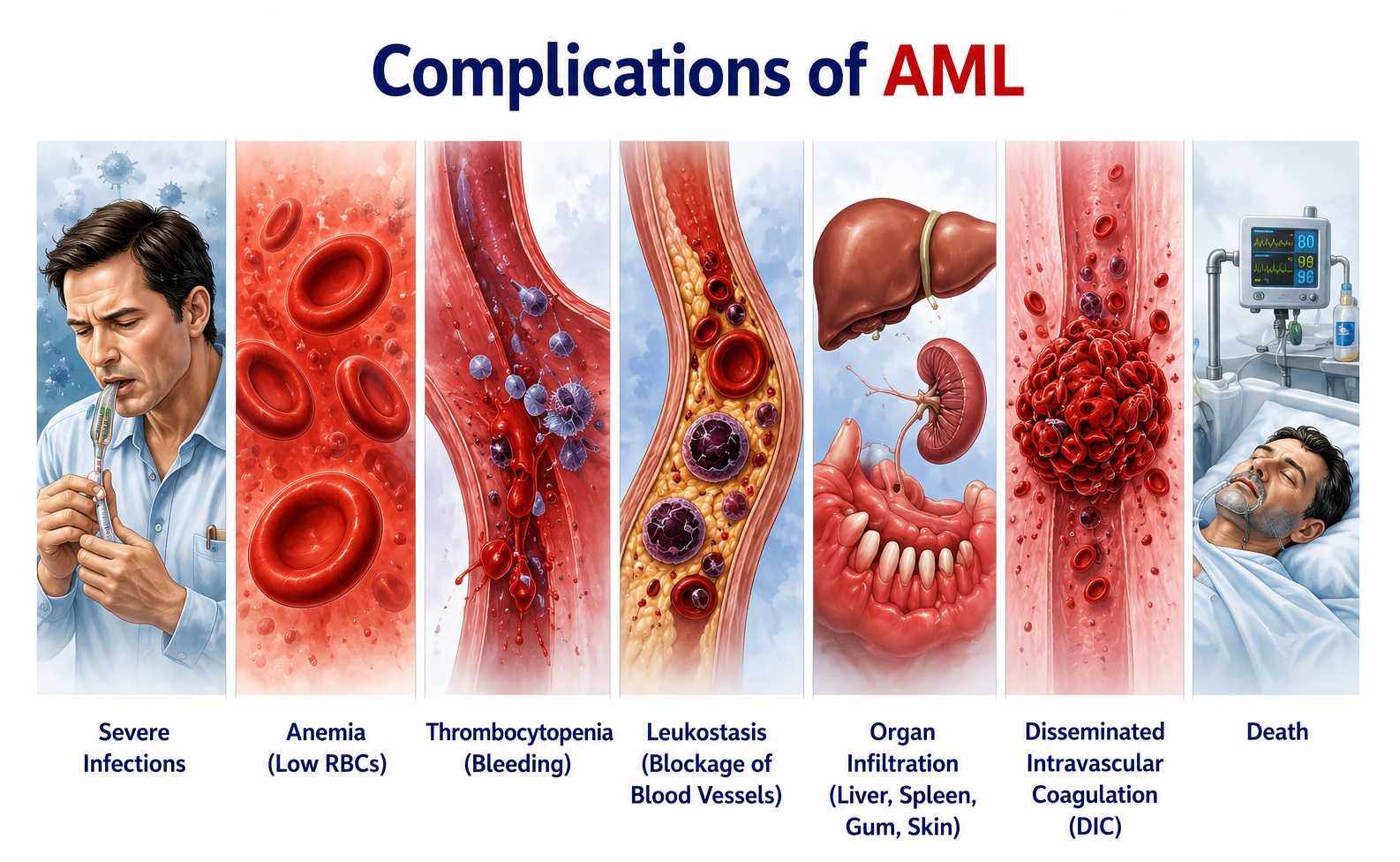
3. Leukostasis
In patients with WBC >100 × 10⁹/L, circulating blasts aggregate in microvasculature causing tissue ischemia. Clinical manifestations include hypoxemic respiratory failure, cerebral dysfunction, and retinal hemorrhages. Emergency management involves cytoreduction with hydroxyurea, leukapheresis in selected cases, and prompt initiation of induction chemotherapy. Caution: red cell transfusion should be withheld until cytoreduction is achieved to avoid further hyperviscosity.
4. Tumor Lysis Syndrome (TLS)
Spontaneous or treatment-induced lysis of leukemic blasts releases massive amounts of intracellular contents. Metabolic consequences include hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia. Acute kidney injury from uric acid and calcium-phosphate precipitation in renal tubules, cardiac arrhythmias from hyperkalemia, and seizures from hypocalcemia may result. Prevention with allopurinol/rasburicase, aggressive intravenous hydration, urinary alkalinization, and electrolyte monitoring is essential.
5. Intracranial Hemorrhage
Life-threatening hemorrhage into the CNS may result from severe thrombocytopenia, DIC, or leukostasis-induced vascular rupture. Presents with sudden severe headache, vomiting, altered consciousness, and focal neurological deficits. Requires emergent neuroimaging, platelet and coagulation factor replacement, and neurosurgical consultation.
6. Relapsed/Refractory AML
Approximately 40–60% of patients who achieve complete remission will eventually relapse. Primary refractory AML (failure to achieve CR after induction) and relapsed AML represent major therapeutic challenges associated with clonal evolution, acquisition of resistance mutations (e.g., FLT3 mutations, TP53 mutations), and poor prognosis. Salvage chemotherapy regimens (FLAG-IDA, MEC, CLAG-M) combined with targeted agents and allogeneic SCT in eligible patients represent the main therapeutic strategies.
B. Treatment-Related Complications
1. Anthracycline-Related Cardiotoxicity
Doxorubicin and idarubicin cause cumulative dose-dependent cardiomyopathy resulting from oxidative myocardial damage. Acute cardiotoxicity manifests as arrhythmias; chronic cardiomyopathy presents as dilated cardiomyopathy with reduced LVEF, potentially progressing to heart failure. Monitoring with serial echocardiography and limiting cumulative anthracycline doses are essential preventive measures.
2. Mucositis and Gastrointestinal Toxicity
High-dose cytarabine and intensive chemotherapy cause diffuse mucositis of the gastrointestinal tract, manifesting as severe oral ulceration, diarrhea, nausea, and vomiting. Severe mucositis impairs oral intake, provides portal-of-entry for bacterial translocation, and may precipitate bacteremia.
3. Neurotoxicity (High-Dose Cytarabine)
High-dose cytarabine (HiDAC ≥3 g/m²), particularly used in consolidation, may cause cerebellar toxicity with gait ataxia, nystagmus, and dysarthria. Renal impairment reduces cytarabine clearance, increasing neurotoxicity risk; dose reduction is required when creatinine clearance is impaired.
4. Differentiation Syndrome (ATRA/ATO-Related)
In APL treated with ATRA ± arsenic trioxide, differentiation of leukemic promyelocytes releases cytokines causing a systemic inflammatory capillary leak syndrome. Manifestations include unexplained fever, dyspnea, hypotension, weight gain, pleural and pericardial effusions, and pulmonary infiltrates. Management requires immediate high-dose dexamethasone (10 mg twice daily) and temporary interruption of ATRA/ATO in severe cases.
5. Transplant-Related Complications (Allo-SCT)
Patients undergoing allogeneic hematopoietic stem cell transplantation face graft-versus-host disease (GvHD — both acute and chronic), transplant-related infections (CMV, EBV, Pneumocystis, Aspergillus), sinusoidal obstruction syndrome (SOS/VOD), engraftment failure, and significant transplant-related mortality — risks balanced against the graft-versus-leukemia effect and potential for cure.
Treatment of Acute Myeloid Leukemia
AML treatment is stratified by age, performance status, cytogenetic/molecular risk, and disease-specific characteristics. The overarching goal in fit patients is to achieve complete remission (CR) with induction therapy and consolidate it with intensive chemotherapy and/or allogeneic stem cell transplantation. In older or unfit patients, less intensive but molecularly targeted regimens are increasingly preferred.
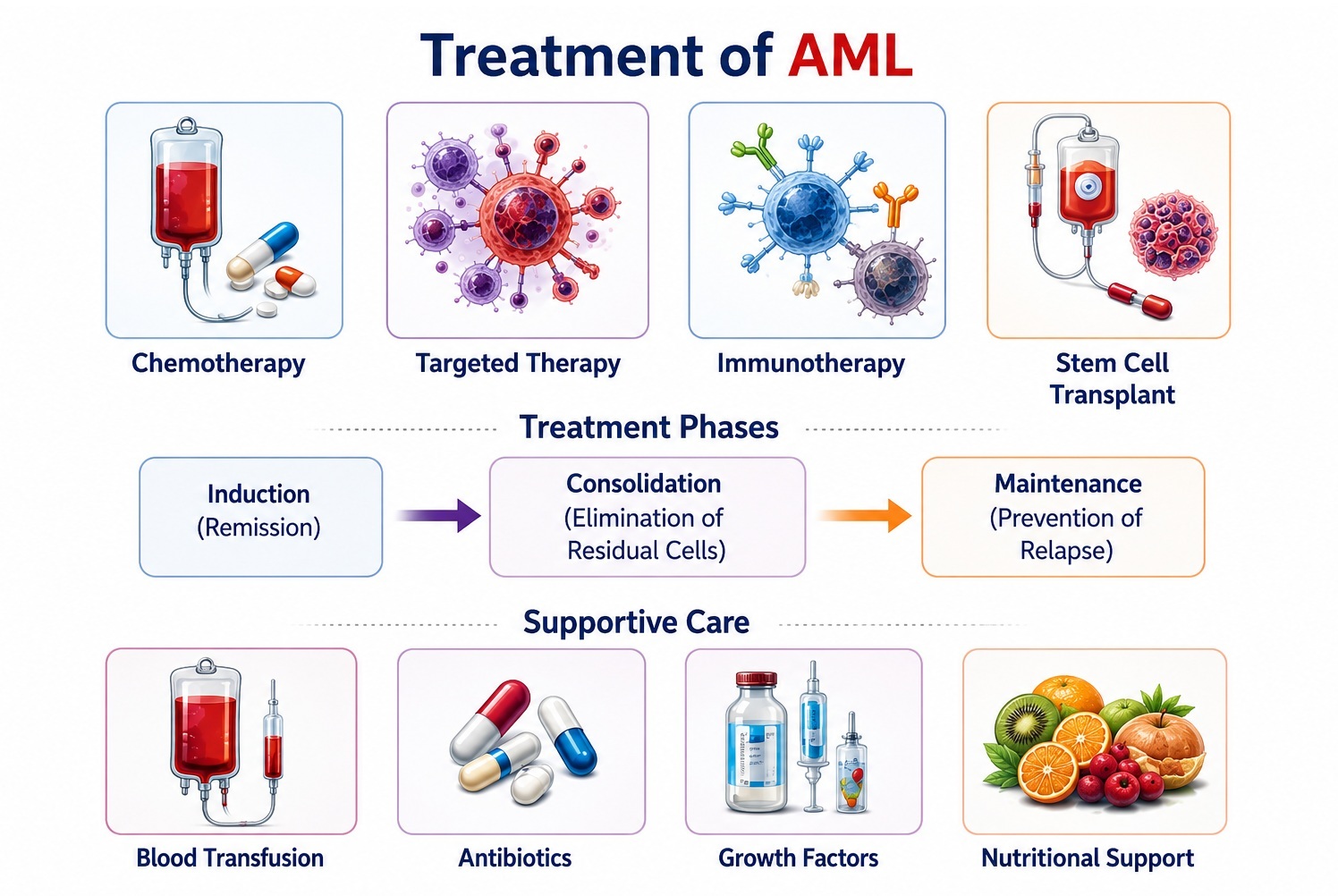
A. Induction Therapy
1. Standard "7+3" Regimen (Fit Patients ≤75 years)
The cornerstone of AML induction for decades, the "7+3" regimen combines continuous infusion cytarabine (100–200 mg/m²/day × 7 days) with an anthracycline (daunorubicin or idarubicin × 3 days). It achieves complete remission in approximately 60–80% of younger adults. A second induction cycle ("5+2") may be administered for residual disease at day 14–21 bone marrow assessment.
2. Cytarabine + Daunorubicin + FLT3 Inhibitor (FLT3-Mutated AML)
For patients with FLT3-mutated AML, the addition of midostaurin (50 mg twice daily on days 8–21 of each cycle) to standard 7+3 induction and consolidation followed by maintenance demonstrated significantly improved overall survival in the RATIFY trial and is now the standard of care for FLT3+ AML in fit patients.
3. Venetoclax + Azacitidine or Decitabine (Older/Unfit Patients)
The combination of venetoclax (BCL-2 inhibitor, 400 mg daily) with hypomethylating agents (HMAs — azacitidine or decitabine) has transformed treatment outcomes for older or comorbid patients ineligible for intensive induction. The VIALE-A trial demonstrated superior overall survival and CR rates compared to azacitidine alone, making this regimen the preferred frontline option for this population. Particular benefit is seen in patients with NPM1 mutations or IDH1/2 mutations.
4. APL-Specific Therapy (ATRA + Arsenic Trioxide ± Chemotherapy)
APL represents a distinct therapeutic entity. ATRA (all-trans retinoic acid) induces differentiation of leukemic promyelocytes, while arsenic trioxide (ATO) promotes apoptosis. The combination of ATRA + ATO (APL0406 / ATRA-ATO regimen) is curative in approximately 95% of low-to-intermediate risk APL patients without requiring cytotoxic chemotherapy — representing the most successful targeted treatment in acute leukemia. High-risk APL (WBC ≥10 × 10⁹/L) still requires chemotherapy-based consolidation.
B. Consolidation Therapy
1. High-Dose Cytarabine (HiDAC)
For core-binding factor (CBF) AML [t(8;21) or inv(16)] with favorable prognosis, 3–4 cycles of HiDAC (3 g/m²/12h × 6 doses per cycle) consolidation following induction CR provide high rates of long-term cure without transplantation. CBF-AML patients benefit substantially from cytarabine-based consolidation, and transplantation in CR1 is not recommended unless MRD positive or refractory.
2. Allogeneic Hematopoietic Stem Cell Transplantation (Allo-SCT)
Allo-SCT provides curative potential through the graft-versus-leukemia (GvL) effect mediated by donor T-lymphocytes. It is the preferred consolidation strategy for:
- All adverse-risk AML patients in first complete remission (CR1) who have a suitable donor
- Intermediate-risk AML patients in CR1, particularly those with FLT3-ITD (high allelic ratio), FLT3+/NPM1-wild type, or MRD positivity after consolidation
- All AML patients in second complete remission (CR2) after relapse
Donor sources include HLA-matched sibling, matched unrelated donor (MUD), haploidentical (half-match) donors, or umbilical cord blood. HLA-matched donors are preferred; haploidentical transplantation with post-transplant cyclophosphamide (PT-Cy) has expanded donor availability significantly.
3. Autologous SCT
Autologous SCT is rarely used in AML but may be considered in selected favorable-risk patients without a suitable allogeneic donor who achieve MRD-negative CR.
C. Targeted and Novel Therapies
1. IDH Inhibitors
- Ivosidenib (IDH1 inhibitor): Approved for IDH1-mutated AML in newly diagnosed (older/unfit patients), relapsed/refractory settings, and in combination with azacitidine.
- Enasidenib and olutasidenib (IDH2 inhibitors): Approved for relapsed/refractory IDH2-mutated AML; olutasidenib also approved for IDH1-mutated AML.
2. FLT3 Inhibitors
- Midostaurin: First-generation FLT3 inhibitor approved as induction/consolidation in FLT3+ AML.
- Gilteritinib: Second-generation selective FLT3 inhibitor approved for relapsed/refractory FLT3-mutated AML; also studied in maintenance post-transplant.
- Quizartinib: Highly selective FLT3-ITD inhibitor approved in combination with standard chemotherapy for newly diagnosed FLT3-ITD–positive AML.
3. Gemtuzumab Ozogamicin (GO)
GO is an anti-CD33 antibody-drug conjugate that delivers calicheamicin to CD33-positive blasts. It is approved for CD33+ AML in combination with standard chemotherapy for newly diagnosed patients (particularly favorable and intermediate risk), and as monotherapy in relapsed CD33+ AML. Risk of hepatic sinusoidal obstruction syndrome (SOS/VOD) requires careful dose monitoring.
4. CPX-351 (Liposomal Cytarabine-Daunorubicin)
A liposomal encapsulated 5:1 molar ratio of cytarabine:daunorubicin, CPX-351 is approved for therapy-related AML and AML with myelodysplasia-related changes (AML-MRC) in adults, where it demonstrates superior survival compared to conventional 7+3 induction.
D. Supportive Care
- Red blood cell transfusion: Maintain hemoglobin ≥7–8 g/dL (higher threshold in elderly or symptomatic patients).
- Platelet transfusion: Prophylactic transfusion at platelet count <10 × 10⁹/L; higher thresholds (<20–50 × 10⁹/L) in patients with active bleeding, fever, or coagulopathy.
- Antimicrobial prophylaxis: Antibacterial (fluoroquinolones), antifungal (azoles, echinocandins), and antiviral (acyclovir) prophylaxis during neutropenic phases; PCP prophylaxis (cotrimoxazole) during steroid-intensive regimens.
- Growth factors: Granulocyte colony-stimulating factor (G-CSF) may be used cautiously to shorten the duration of neutropenia post-induction in selected protocols.
- TLS prophylaxis: Allopurinol pre-chemotherapy for standard-risk patients; rasburicase (recombinant urate oxidase) for high-risk patients with elevated uric acid or hyperleukocytosis.
- Nutritional support: Enteral nutrition via NGT or TPN for patients with severe mucositis and inadequate oral intake.
- Palliative care and psychosocial support: Early integration of palliative care services, psychological counseling, and social support is essential throughout the treatment continuum.
Prevention and Risk Reduction in Acute Myeloid Leukemia
AML arising de novo cannot currently be prevented in the majority of cases, as most cases develop sporadically without identifiable preventable antecedents. However, several evidence-based strategies can reduce risk, facilitate early detection of pre-leukemic states, and enable timely intervention:
A. Avoidance of Modifiable Environmental Exposures
- Benzene avoidance: Occupational safety regulations limiting benzene exposure in petroleum, chemical, and manufacturing industries are critical. Workers should comply with personal protective equipment requirements, ventilation standards, and regular biological monitoring of benzene metabolites.
- Tobacco cessation: Smoking cessation reduces AML risk attributable to cigarette smoke-derived benzene and carcinogens. Public health tobacco control measures have population-level benefits on leukemia incidence.
- Pesticide and herbicide regulation: Minimizing occupational and residential exposure to recognized hematopoietic carcinogens through safe agricultural practices and regulatory oversight.
- Radiation safety: Adherence to ALARA (As Low As Reasonably Achievable) principles in medical imaging, occupational radiation monitoring, and avoidance of unnecessary exposure to ionizing radiation.
B. Surveillance and Early Detection of Pre-Leukemic States
- Monitoring of MDS and MPNs: Regular bone marrow assessments in high-grade MDS (RAEB) and MPN patients to detect early transformation to AML, enabling timely consideration of hypomethylating agents or stem cell transplantation before overt transformation.
- Clonal hematopoiesis of indeterminate potential (CHIP): Although no proven intervention modifies CHIP progression risk, awareness among primary care providers and hematologists of CHIP as a pre-malignant state enables risk-stratified monitoring, particularly in individuals with cardiovascular comorbidities where concomitant management may be warranted.
- Hereditary predisposition syndromes: Genetic counseling and surveillance programs for families with germline RUNX1, CEBPA, DDX41, GATA2, or ETV6 mutations allow early detection of hematological abnormalities and informed decision-making regarding prophylactic stem cell transplantation in selected cases.
C. Prevention in Therapy-Related AML
- Optimize cytotoxic therapy regimens: For cancer survivors treated with alkylating agents or topoisomerase II inhibitors, use of the lowest effective cumulative doses and exploration of non-cytotoxic alternatives or targeted therapies where feasible may reduce therapy-related AML risk.
- Long-term cancer survivorship surveillance: CBC monitoring in patients who have received significant cytotoxic chemotherapy facilitates early detection of cytopenias, dysplastic changes, or AML.
- Informed consent in oncology: Patients receiving potentially leukemogenic chemotherapy must receive thorough counseling regarding the risk of secondary malignancy.
D. Infection Prevention and Supportive Prevention
- Neutropenic precautions: Strict hand hygiene, avoidance of contact with sick individuals, safe food handling, and low-microbial diet (avoiding raw fruits, vegetables, unpasteurized products) during neutropenic periods reduce infectious complications.
- Vaccination: Inactivated vaccines (influenza, pneumococcal, COVID-19) are recommended for AML patients in stable remission. Live vaccines must be avoided in immunosuppressed states.
- Antifungal prophylaxis: Triazole antifungals (posaconazole, voriconazole) during intensive induction and consolidation chemotherapy reduce invasive aspergillosis incidence and mortality in high-risk patients.
E. Research and Future Preventive Strategies
- Cancer genome surveillance: Population-based genomic screening platforms may in the future identify clonal hematopoiesis or pre-AML mutations in asymptomatic individuals, enabling chemopreventive interventions to delay or prevent overt leukemia — currently under active research investigation.
- Immunotherapy and cancer vaccines: Peptide-based leukemia vaccines targeting mutant antigens (Wilms tumor antigen WT-1, PR1, NPM1 neoepitopes) are being investigated for AML prevention and post-remission maintenance.
- Targeting inflammation: Chronic inflammation promotes clonal evolution; anti-inflammatory strategies targeting IL-1β and IL-6 pathways are being explored in pre-leukemic conditions including high-risk MDS and CHIP.
Common FAQs on Acute Myeloid Leukemia
Bibliography on Acute Myeloid Leukemia
- Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 ELN recommendations from an international expert panel. Blood. 2022;140(12):1345-1377.
- Harrison's Principles of Internal Medicine, 21st ed. Editors: Jameson JL, Fauci AS, Kasper DL, et al. McGraw-Hill Education.
- Williams Hematology, 10th ed. Editors: Kaushansky K, Prchal JT, Burns LJ, et al. McGraw-Hill Education.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations. Blood. 2017;129(4):424-447.
- DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629. [VIALE-A trial]
- Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. [RATIFY trial]
- Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
- DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398.
- Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111-121. [APL0406 trial]
- Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary AML. J Clin Oncol. 2018;36(26):2684-2692.
- Kayser S, Döhner K, Krauter J, et al. Outcome of AML with gemtuzumab ozogamicin consolidation: results from the randomized AML2003 trial. Leukemia. 2013;27:1:26-33.
- WHO Classification of Tumours Editorial Board. WHO Classification of Haematolymphoid Tumours, 5th ed. IARC Press, Lyon. 2022.
- Wintrobe's Clinical Hematology, 14th ed. Editors: Greer JP, Arber DA, Glader BE, et al. Lippincott Williams & Wilkins.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
- Hematology: Basic Principles and Practice, 8th ed. Editors: Hoffman R, Benz EJ Jr, Silberstein LE, et al. Elsevier.